Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, 90095, USA.
Medical Informatics Home Area, Department of Bioinformatics, University of California, Los Angeles, Los Angeles, CA, 90024, USA.
Commun Biol. 2024 Aug 25;7(1):1049. doi: 10.1038/s42003-024-06742-0.
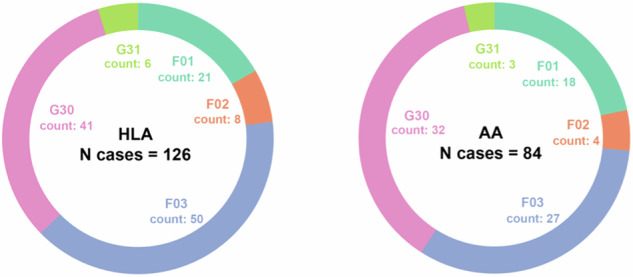
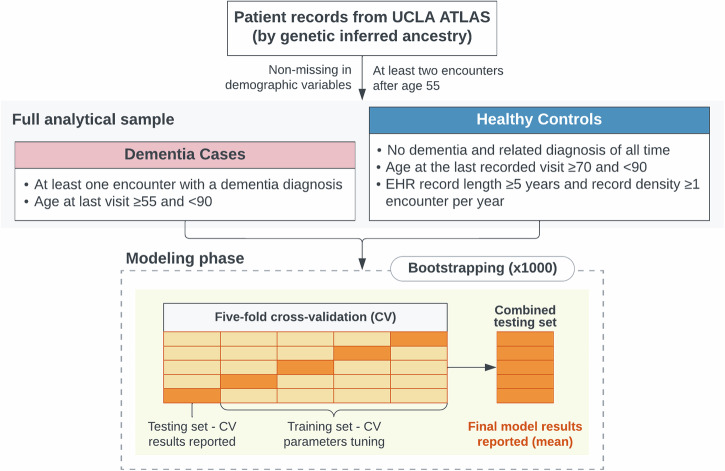
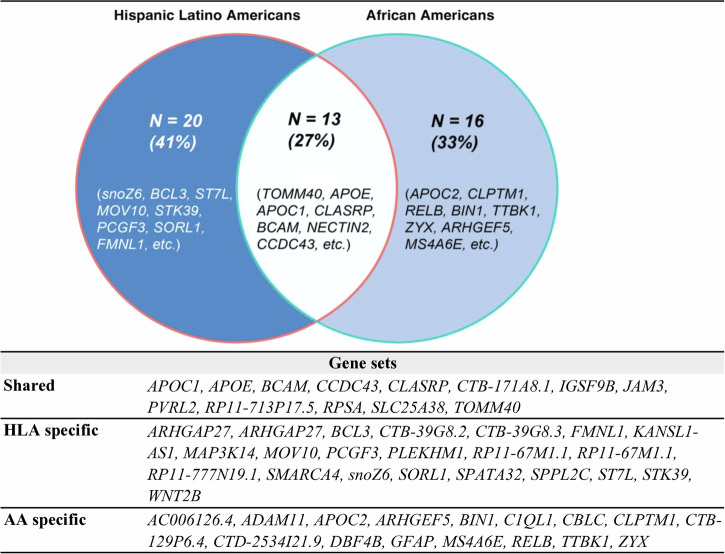
Genetic risk modeling for dementia offers significant benefits, but studies based on real-world data, particularly for underrepresented populations, are limited. We employ an Elastic Net model for dementia risk prediction using single-nucleotide polymorphisms prioritized by functional genomic data from multiple neurodegenerative disease genome-wide association studies. We compare this model with APOE and polygenic risk score models across genetic ancestry groups (Hispanic Latino American sample: 610 patients with 126 cases; African American sample: 440 patients with 84 cases; East Asian American sample: 673 patients with 75 cases), using electronic health records from UCLA Health for discovery and the All of Us cohort for validation. Our model significantly outperforms other models across multiple ancestries, improving the area-under-precision-recall curve by 31-84% (Wilcoxon signed-rank test p-value <0.05) and the area-under-the-receiver-operating characteristic by 11-17% (DeLong test p-value <0.05) compared to the APOE and the polygenic risk score models. We identify shared and ancestry-specific risk genes and biological pathways, reinforcing and adding to existing knowledge. Our study highlights the benefits of integrating functional mapping, multiple neurodegenerative diseases, and machine learning for genetic risk models in diverse populations. Our findings hold potential for refining precision medicine strategies in dementia diagnosis.
基于人群的痴呆遗传风险建模具有重要意义,但基于真实世界数据的研究,特别是针对代表性不足人群的研究还很有限。我们使用弹性网络模型,基于来自多个神经退行性疾病全基因组关联研究的功能基因组数据优先考虑单核苷酸多态性,对痴呆风险进行预测。我们将该模型与 APOE 和多基因风险评分模型在遗传血统群体(西班牙裔拉丁裔美国样本:610 名患者中有 126 例;非裔美国人样本:440 名患者中有 84 例;东亚裔美国人样本:673 名患者中有 75 例)中进行比较,使用来自加州大学洛杉矶分校健康电子病历进行发现,使用全美国人队列进行验证。我们的模型在多个血统中均显著优于其他模型,与 APOE 和多基因风险评分模型相比,在精度-召回曲线下面积方面提高了 31-84%(Wilcoxon 符号秩检验 p 值<0.05),在接收者操作特征曲线下面积方面提高了 11-17%(DeLong 检验 p 值<0.05)。我们确定了共享和血统特异性的风险基因和生物学途径,强化和补充了现有知识。我们的研究强调了整合功能映射、多种神经退行性疾病和机器学习在不同人群中的遗传风险模型中的优势。我们的研究结果为痴呆症诊断中精准医学策略的改进提供了潜力。