Institute of Cytology and Genetics, Siberian Branch of Russian Academy of Sciences, Ave. Lavrentiev, 10, 630090 Novosibirsk, Russia.
Institute of General Genetics, Russian Academy of Sciences, Gubkin St. 3, 119311 Moscow, Russia.
Genes (Basel). 2024 Sep 7;15(9):1174. doi: 10.3390/genes15091174.
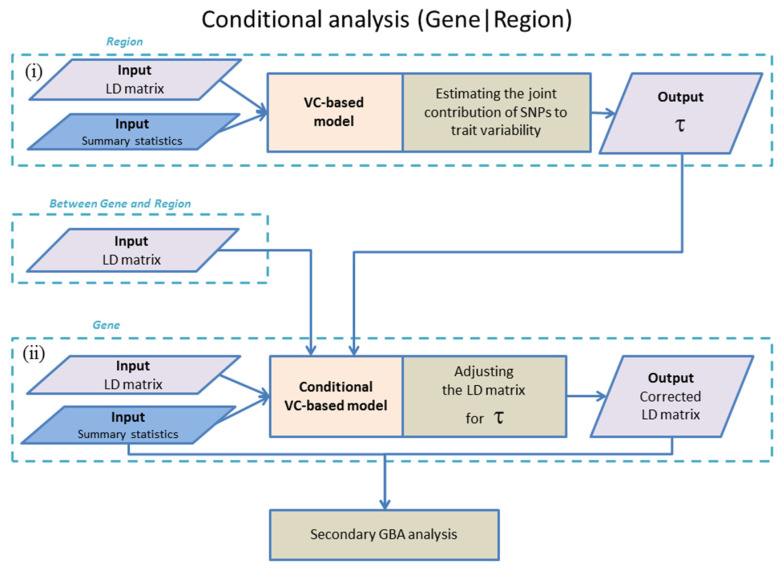
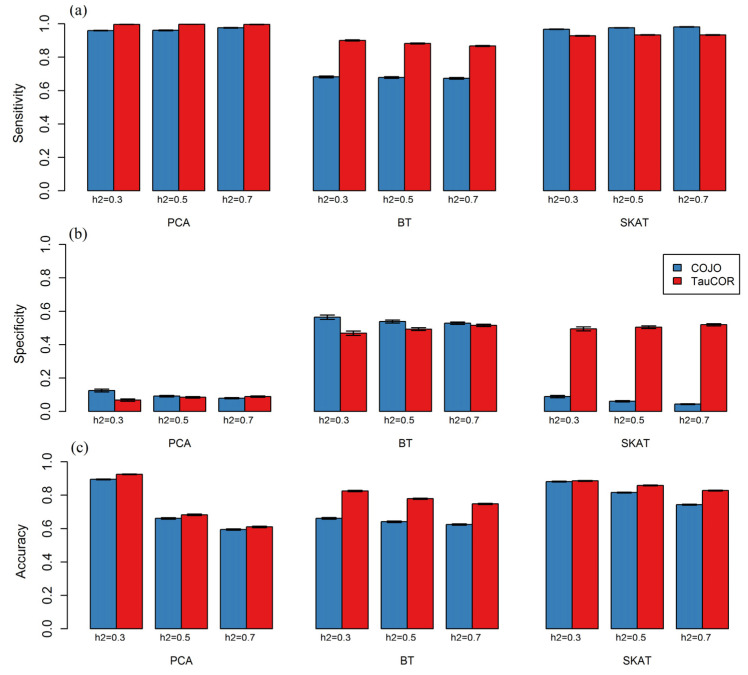
Gene-based association analysis is a powerful tool for identifying genes that explain trait variability. An essential step of this analysis is a conditional analysis. It aims to eliminate the influence of SNPs outside the gene, which are in linkage disequilibrium with intragenic SNPs. The popular conditional analysis method, GCTA-COJO, accounts for the influence of several top independently associated SNPs outside the gene, correcting the z statistics for intragenic SNPs. We suggest a new TauCOR method for conditional gene-based analysis using summary statistics. This method accounts the influence of the full regional polygenic background, correcting the genotype correlations between intragenic SNPs. As a result, the distribution of z statistics for intragenic SNPs becomes conditionally independent of distribution for extragenic SNPs. TauCOR is compatible with any gene-based association test. TauCOR was tested on summary statistics simulated under different scenarios and on real summary statistics for a 'gold standard' gene list from the Open Targets Genetics project. TauCOR proved to be effective in all modelling scenarios and on real data. The TauCOR's strategy showed comparable sensitivity and higher specificity and accuracy than GCTA-COJO on both simulated and real data. The method can be successfully used to improve the effectiveness of gene-based association analyses.
基于基因的关联分析是识别解释性状变异的基因的有力工具。该分析的一个重要步骤是条件分析。它旨在消除与基因内 SNP 处于连锁不平衡的基因外 SNP 的影响。流行的条件分析方法 GCTA-COJO 考虑了基因外几个最重要的独立关联 SNP 的影响,校正了基因内 SNP 的 z 统计量。我们建议使用汇总统计数据为条件基因基础分析使用新的 TauCOR 方法。该方法考虑了全区域多基因背景的影响,校正了基因内 SNP 之间的基因型相关性。因此,基因内 SNP 的 z 统计量的分布条件独立于基因外 SNP 的分布。TauCOR 与任何基于基因的关联测试都兼容。TauCOR 在不同情况下模拟的汇总统计数据和来自 Open Targets Genetics 项目的“黄金标准”基因列表的真实汇总统计数据上进行了测试。TauCOR 在所有建模场景和真实数据上都被证明是有效的。在模拟和真实数据上,TauCOR 的策略与 GCTA-COJO 相比,均显示出可比的灵敏度和更高的特异性和准确性。该方法可成功用于提高基于基因的关联分析的有效性。