Xu Xianjin, Kao Wei-Ling, Wang Allison, Lee Hsin-Jou, Duan Rui, Holmes Hannah, Gallazzi Fabio, Ji Juan, Sun Hongmin, Heng Xiao, Zou Xiaoqin
Department of Physics, University of Missouri, Columbia, MO 65211, USA.
Department of Biochemistry, University of Missouri, Columbia, MO 65211, USA.
PNAS Nexus. 2024 Nov 27;3(12):pgae541. doi: 10.1093/pnasnexus/pgae541. eCollection 2024 Dec.
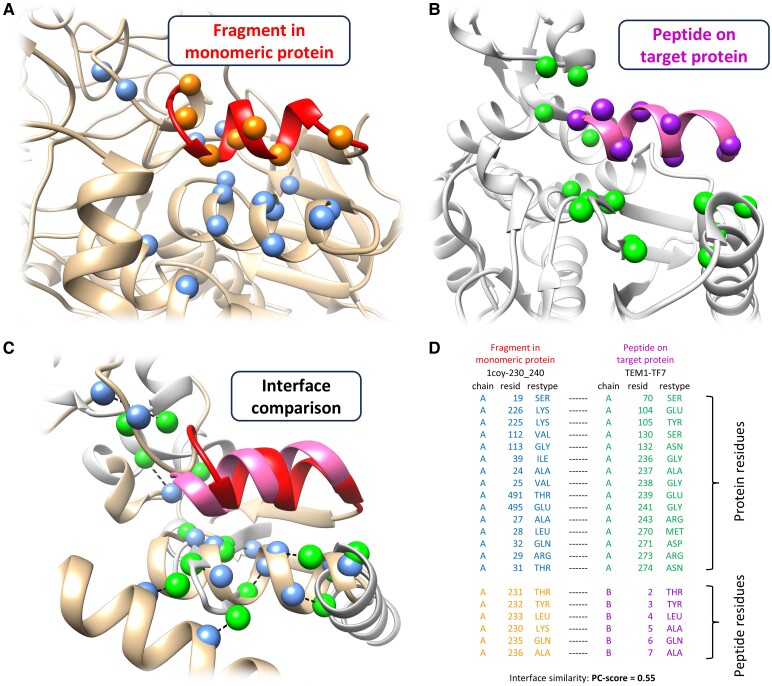
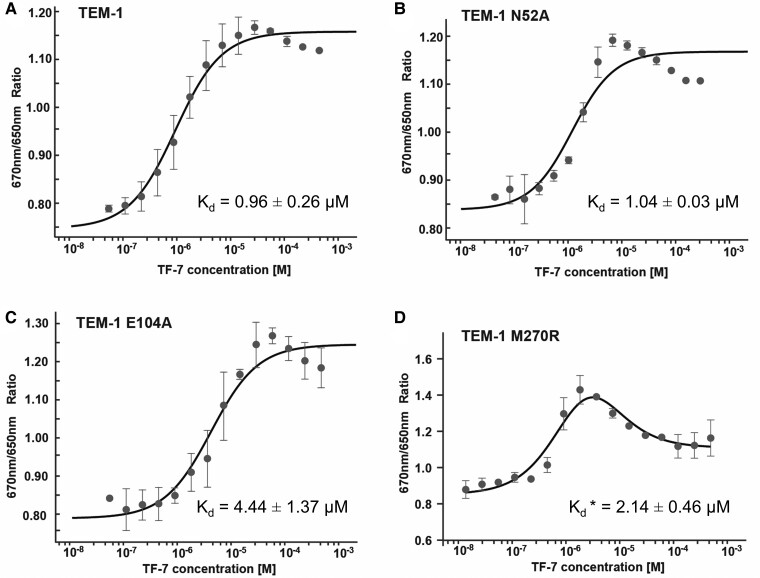
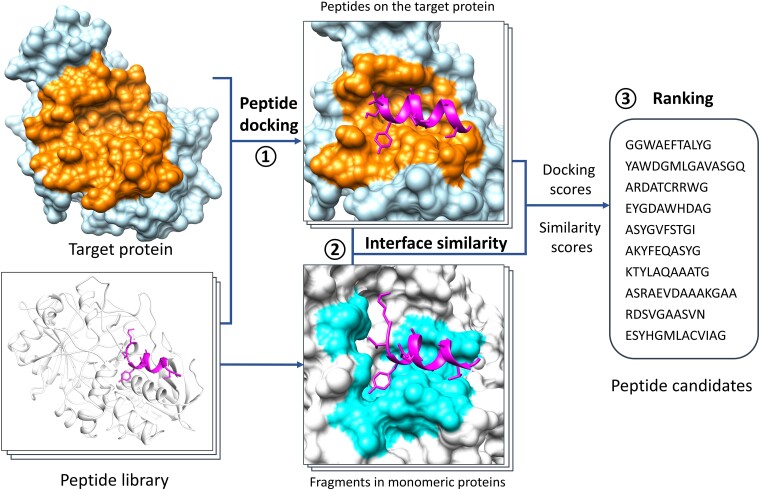
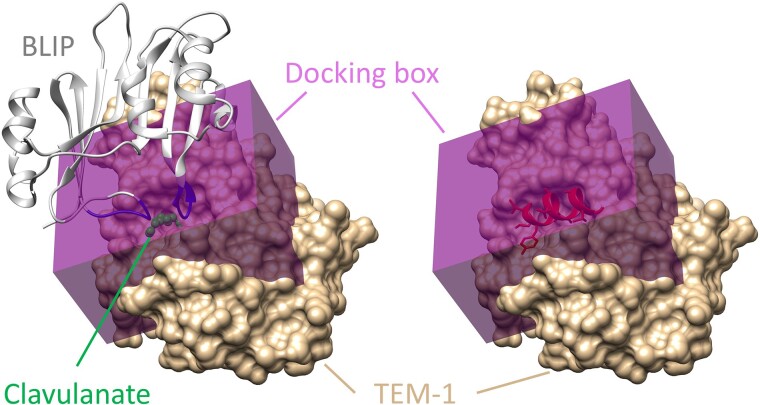
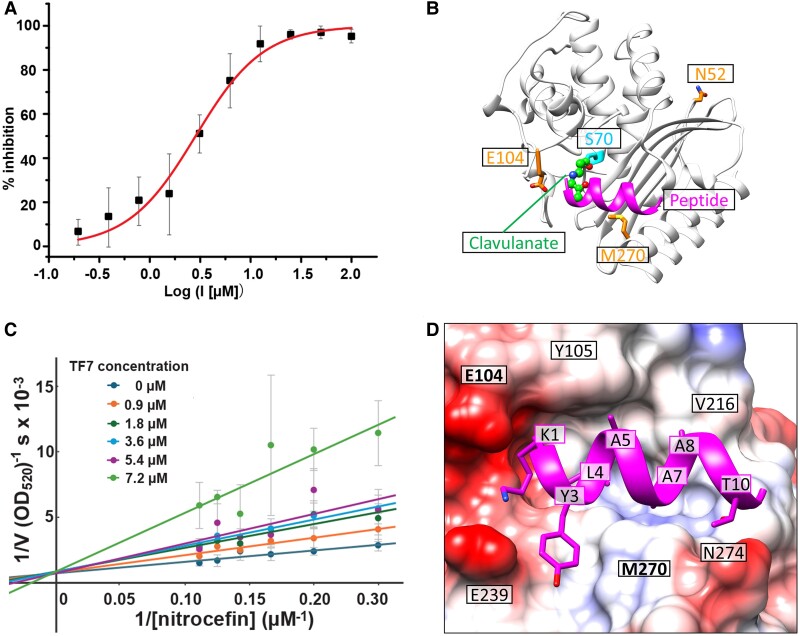
The field of therapeutic peptides is experiencing a surge, fueled by their advantageous features. These include predictable metabolism, enhanced safety profile, high selectivity, and reduced off-target effects compared with small-molecule drugs. Despite progress in addressing limitations associated with peptide drugs, a significant bottleneck remains: the absence of a large-scale in silico screening method for a given protein target structure. Such methods have proven invaluable in accelerating small-molecule drug discovery. The high flexibility of peptide structures and the large diversity of peptide sequences greatly hinder the development of urgently needed computational methods. Here, we report a method called MDockPeP2_VS to address these challenges. It integrates molecular docking with structural conservation between protein folding and protein-peptide binding. Briefly, we discovered that when the interfacial residues are conserved, a sequence fragment derived from a monomeric protein exhibits a high propensity to bind a target protein with a similar conformation. This valuable insight significantly reduces the search space for peptide conformations, resulting in a substantial reduction in computational time and making in silico peptide screening practical. We applied MDockPeP2_VS to develop peptide inhibitors targeting the TEM-1 β-lactamase of , a key mechanism behind antibiotic resistance in gram-negative bacteria. Among the top 10 peptides selected from in silico screening, TF7 (KTYLAQAAATG) showed significant inhibition of β-lactamase activity with a value of 1.37 ± 0.37 µM. This fully automated, large-scale structure-based in silico peptide screening software is available for free download at https://zougrouptoolkit.missouri.edu/mdockpep2_vs/download.html.
治疗性肽领域正经历着蓬勃发展,这得益于其诸多优势特性。与小分子药物相比,这些特性包括可预测的代谢、更高的安全性、高选择性以及更低的脱靶效应。尽管在解决与肽类药物相关的局限性方面取得了进展,但一个重大瓶颈仍然存在:缺乏针对给定蛋白质靶标结构的大规模计算机模拟筛选方法。事实证明,此类方法在加速小分子药物研发方面具有不可估量的价值。肽结构的高度灵活性和肽序列的巨大多样性极大地阻碍了急需的计算方法的开发。在此,我们报告一种名为MDockPeP2_VS的方法来应对这些挑战。它将分子对接与蛋白质折叠和蛋白质 - 肽结合之间的结构保守性相结合。简而言之,我们发现当界面残基保守时,源自单体蛋白质的序列片段具有以相似构象结合靶蛋白的高度倾向。这一有价值的见解显著减少了肽构象的搜索空间,从而大幅缩短了计算时间,并使计算机模拟肽筛选变得切实可行。我们应用MDockPeP2_VS来开发针对革兰氏阴性菌抗生素耐药性关键机制——TEM - 1β - 内酰胺酶的肽抑制剂。在从计算机模拟筛选中选出的前10种肽中,TF7(KTYLAQAAATG)对β - 内酰胺酶活性表现出显著抑制作用,IC50值为1.37±0.37 μM。这款完全自动化的、基于结构的大规模计算机模拟肽筛选软件可在https://zougrouptoolkit.missouri.edu/mdockpep2_vs/download.html免费下载。