Su Lei, Guo Jindan, Shi Weixiong, Tong Wei, Li Xue, Yang Bochao, Xiang Zhiguang, Qin Chuan
NHC Key Laboratory of Human Disease Comparative Medicine, National Human Diseases Animal Model Resource Center, International Center for Technology and Innovation of Animal Model, Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences (CAMS) & Comparative Medicine Center, Peking Union Medical College (PUMC), Beijing, 100021, China.
BMC Microbiol. 2024 Dec 19;24(1):530. doi: 10.1186/s12866-024-03696-5.
The intestinal microbiota plays a crucial role in health and disease. This study aimed to assess the composition and functional diversity of the intestinal microbiota in donkeys and cows by examining samples collected from different segments of the digestive tract using two distinct techniques: direct swab sampling and faecal sampling.
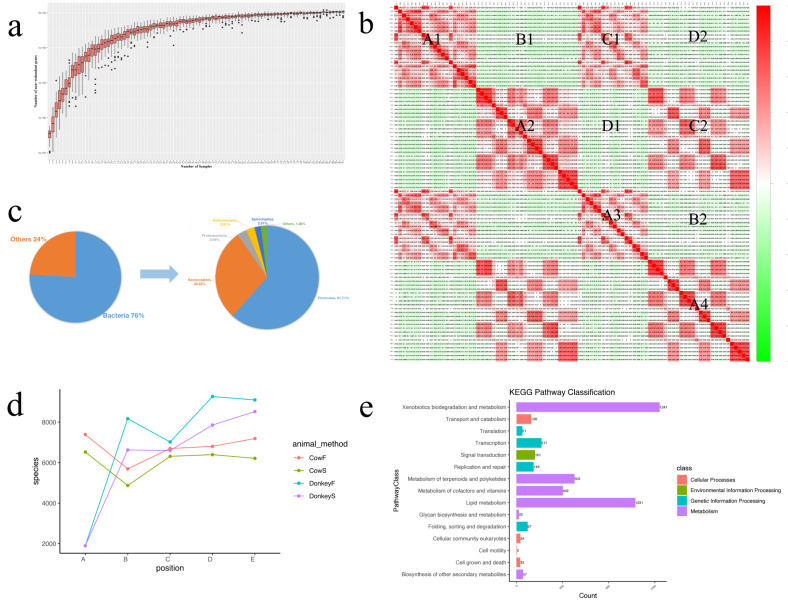
In this study, we investigated and compared the effects of multiple factors on the composition and function of the intestinal microbial community. Approximately 300 GB of metagenomic sequencing data from 91 samples obtained from various segments of the digestive tract were used, including swabs and faecal samples from monogastric animals (donkeys) and polygastric animals (cows). We assembled 4,004,115 contigs for cows and 2,938,653 contigs for donkeys, with a total of 9,060,744 genes. Our analysis revealed that, compared with faecal samples, swab samples presented a greater abundance of Bacteroidetes, whereas faecal samples presented a greater abundance of Firmicutes. Additionally, we observed significant variations in microbial composition among different digestive tract segments in both animals. Our study identified key bacterial species and pathways via different methods and provided evidence that multiple factors can influence the microbial composition. These findings provide new insights for the accurate characterization of the composition and function of the gut microbiota in microbiome research.
The results obtained by both sampling methods in the present study revealed that the composition and function of the intestinal microbiota in donkeys and cows exhibit species-specific and region-specific differences. These findings highlight the importance of using standardized sampling protocols to ensure accurate and consistent characterization of the intestinal microbiota in various animal species. The implications and underlying mechanisms of these associations provide multiple perspectives for future microbiome research.
肠道微生物群在健康和疾病中起着至关重要的作用。本研究旨在通过使用两种不同技术(直接拭子采样和粪便采样)检查从消化道不同部位采集的样本,评估驴和牛肠道微生物群的组成和功能多样性。
在本研究中,我们调查并比较了多种因素对肠道微生物群落组成和功能的影响。使用了从消化道各部位获得的91个样本的约300GB宏基因组测序数据,包括来自单胃动物(驴)和多胃动物(牛)的拭子和粪便样本。我们为牛组装了4,004,115个重叠群,为驴组装了2,938,653个重叠群,共有9,060,744个基因。我们的分析表明,与粪便样本相比,拭子样本中拟杆菌门的丰度更高,而粪便样本中厚壁菌门的丰度更高。此外,我们观察到两种动物不同消化道段之间的微生物组成存在显著差异。我们的研究通过不同方法确定了关键细菌物种和途径,并提供了多种因素可影响微生物组成的证据。这些发现为微生物组研究中准确表征肠道微生物群的组成和功能提供了新见解。
本研究中两种采样方法获得的结果表明,驴和牛肠道微生物群的组成和功能存在物种特异性和区域特异性差异。这些发现凸显了使用标准化采样方案以确保准确一致地表征各种动物物种肠道微生物群的重要性。这些关联的影响和潜在机制为未来的微生物组研究提供了多个视角。