Zhang Chenyue, Wang Qinxin, Li Yiyang, Teng Anqi, Hu Gang, Wuyun Qiqige, Zheng Wei
NITFID, School of Statistics and Data Science, LPMC and KLMDASR, Nankai University, Tianjin 300071, China.
Suzhou New & High-Tech Innovation Service Center, Suzhou 215011, China.
Biomolecules. 2024 Nov 29;14(12):1531. doi: 10.3390/biom14121531.
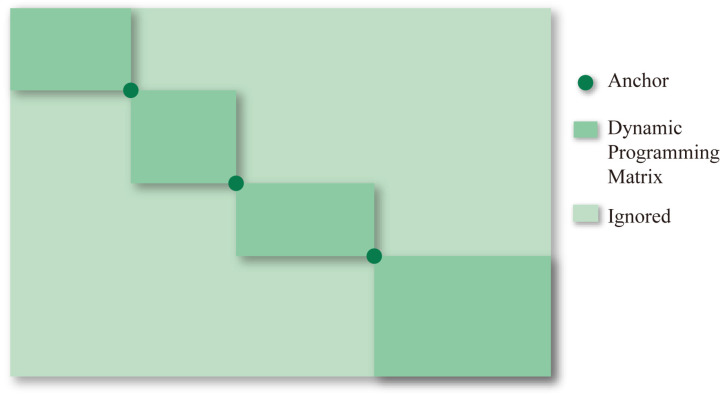
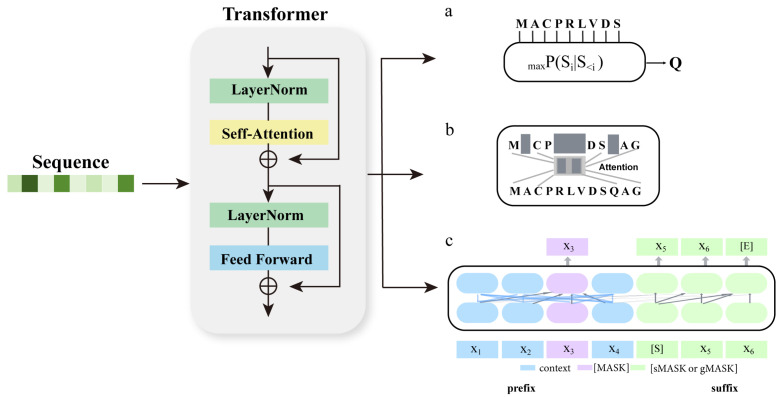
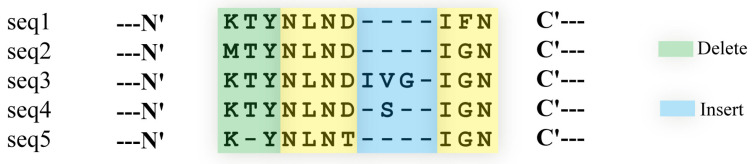
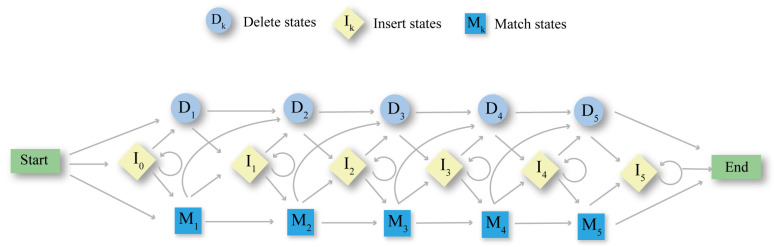
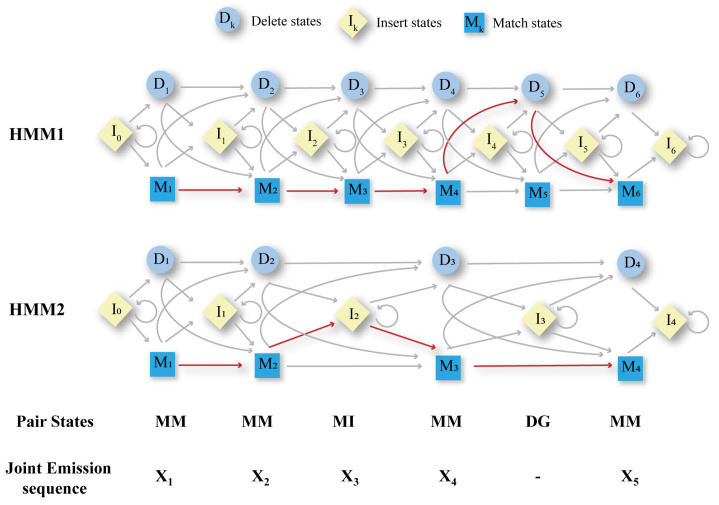
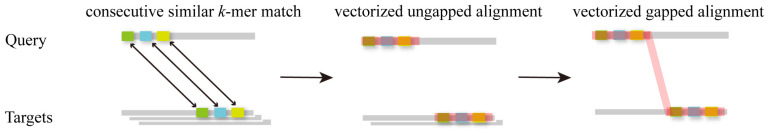
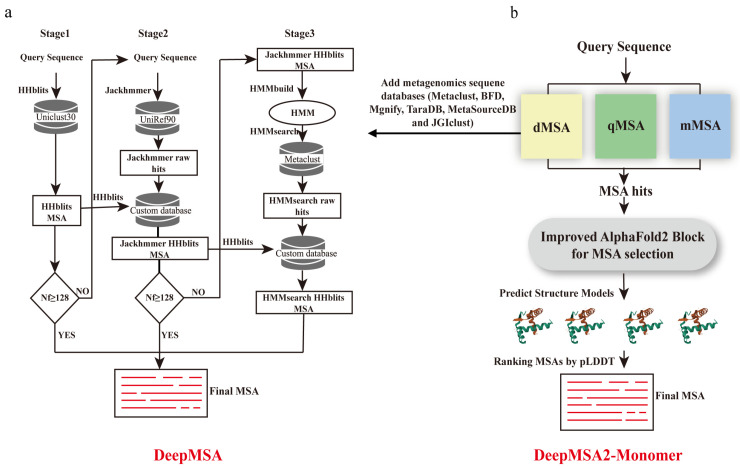
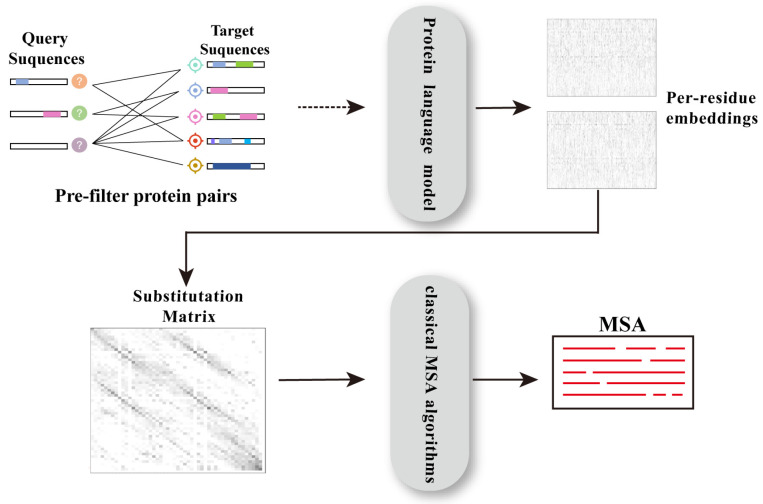
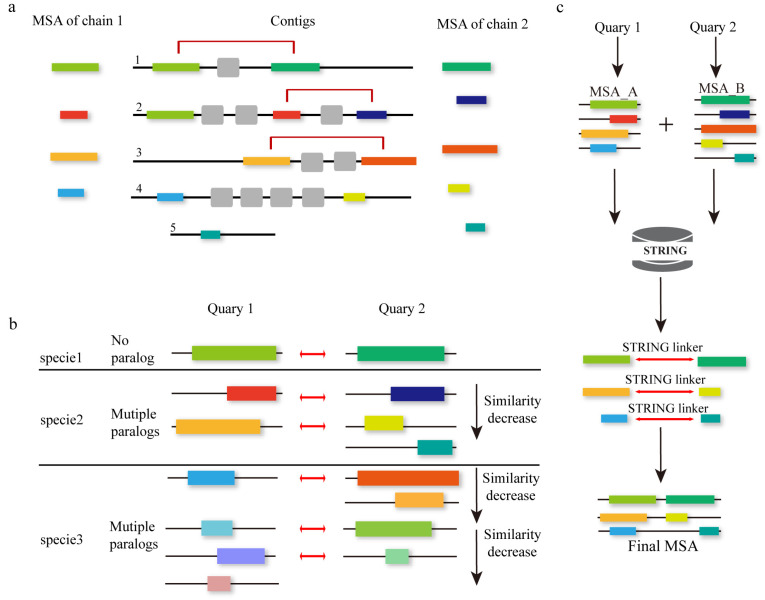
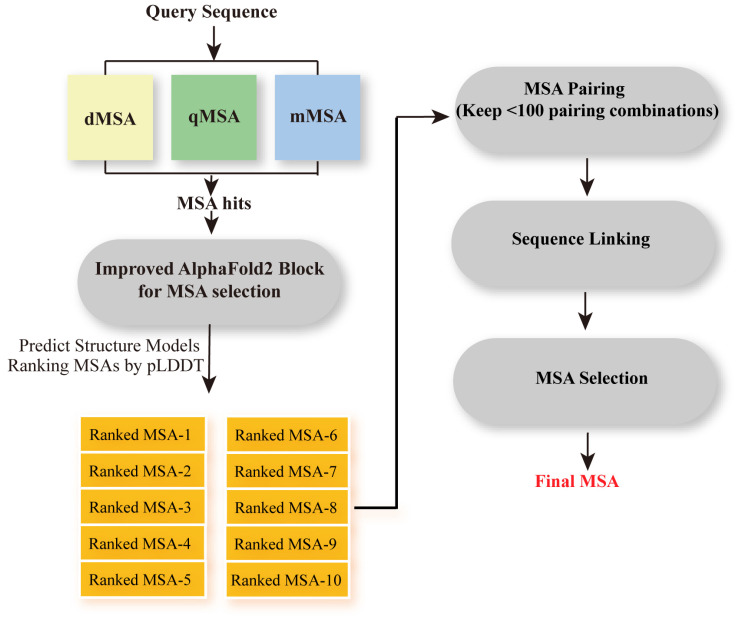
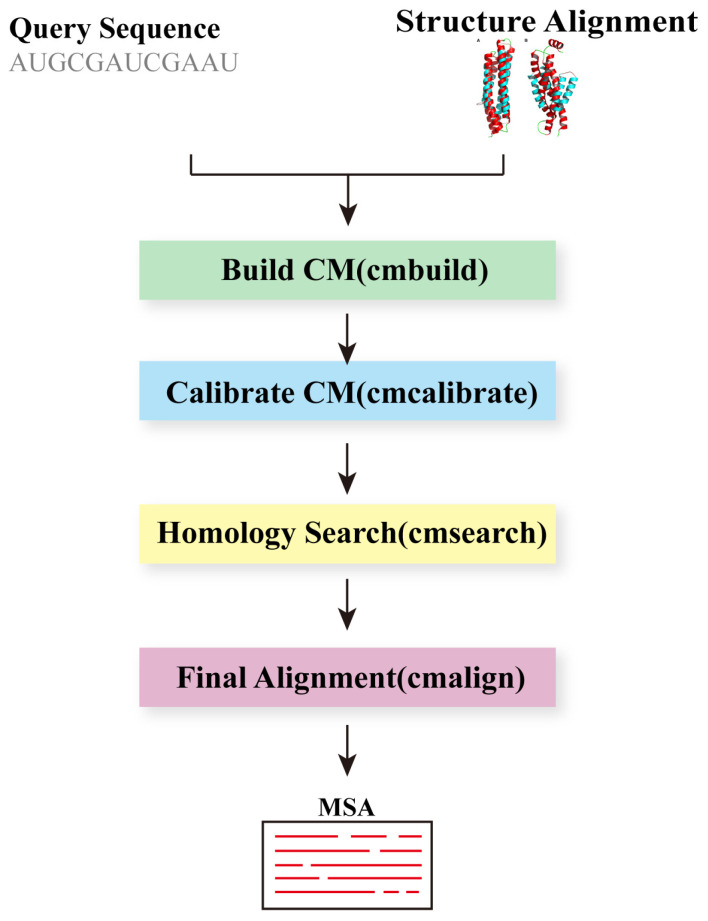
Multiple sequence alignment (MSA) has evolved into a fundamental tool in the biological sciences, playing a pivotal role in predicting molecular structures and functions. With broad applications in protein and nucleic acid modeling, MSAs continue to underpin advancements across a range of disciplines. MSAs are not only foundational for traditional sequence comparison techniques but also increasingly important in the context of artificial intelligence (AI)-driven advancements. Recent breakthroughs in AI, particularly in protein and nucleic acid structure prediction, rely heavily on the accuracy and efficiency of MSAs to enhance remote homology detection and guide spatial restraints. This review traces the historical evolution of MSA, highlighting its significance in molecular structure and function prediction. We cover the methodologies used for protein monomers, protein complexes, and RNA, while also exploring emerging AI-based alternatives, such as protein language models, as complementary or replacement approaches to traditional MSAs in application tasks. By discussing the strengths, limitations, and applications of these methods, this review aims to provide researchers with valuable insights into MSA's evolving role, equipping them to make informed decisions in structural prediction research.
多序列比对(MSA)已发展成为生物科学中的一项基础工具,在预测分子结构和功能方面发挥着关键作用。由于在蛋白质和核酸建模中有着广泛应用,MSA仍然是一系列学科进步的基础。MSA不仅是传统序列比较技术的基础,在人工智能(AI)驱动的进步背景下也日益重要。AI领域的最新突破,尤其是在蛋白质和核酸结构预测方面,在很大程度上依赖于MSA的准确性和效率,以加强远程同源性检测并指导空间限制。本综述追溯了MSA的历史演变,强调了其在分子结构和功能预测中的重要性。我们涵盖了用于蛋白质单体、蛋白质复合物和RNA的方法,同时还探索了新兴的基于AI的替代方法,如蛋白质语言模型,作为传统MSA在应用任务中的补充或替代方法。通过讨论这些方法的优点、局限性和应用,本综述旨在为研究人员提供关于MSA不断演变的作用的宝贵见解,使他们能够在结构预测研究中做出明智的决策。