Liu Xiaofang, Li Bin, Lin Yuxi, Ma Xueying, Liu Yingying, Ma Lili, Ma Xiaomeng, Wang Xia, Li Nanjing, Liu Xiaoyun, Chen Xiaohong
Department of Neurology, The Third Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China.
Department of Infectious Diseases, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China.
Front Mol Biosci. 2025 Jan 7;11:1520050. doi: 10.3389/fmolb.2024.1520050. eCollection 2024.
Emerging evidence underscores the comorbidity mechanisms among autoimmune diseases (AIDs), with innovative technologies such as single-cell RNA sequencing (scRNA-seq) significantly advancing the explorations in this field. This study aimed to investigate the shared genes among three AIDs-Multiple Sclerosis (MS), Systemic Lupus Erythematosus (SLE), and Rheumatoid Arthritis (RA) using bioinformatics databases, and to identify potential biomarkers for early diagnosis.
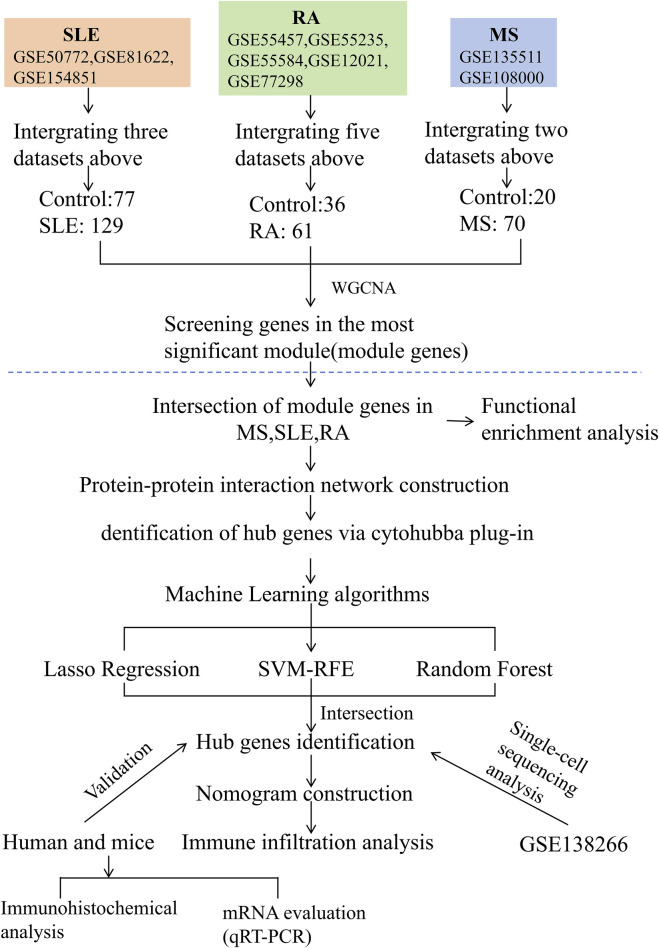
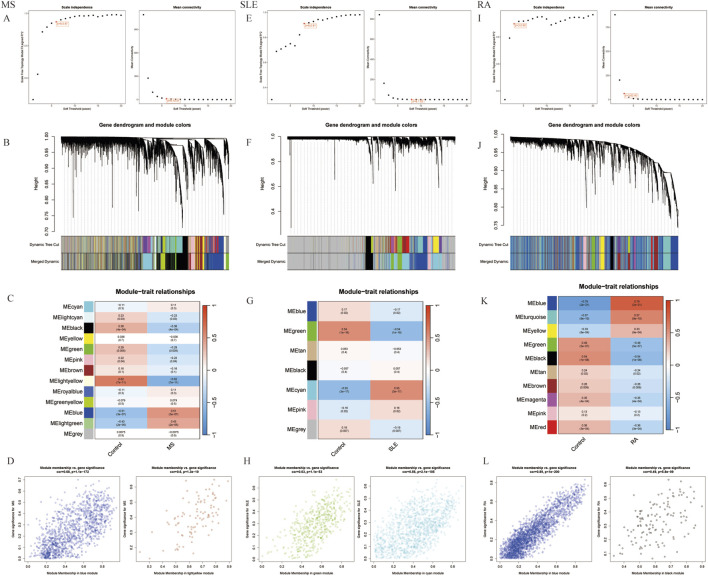
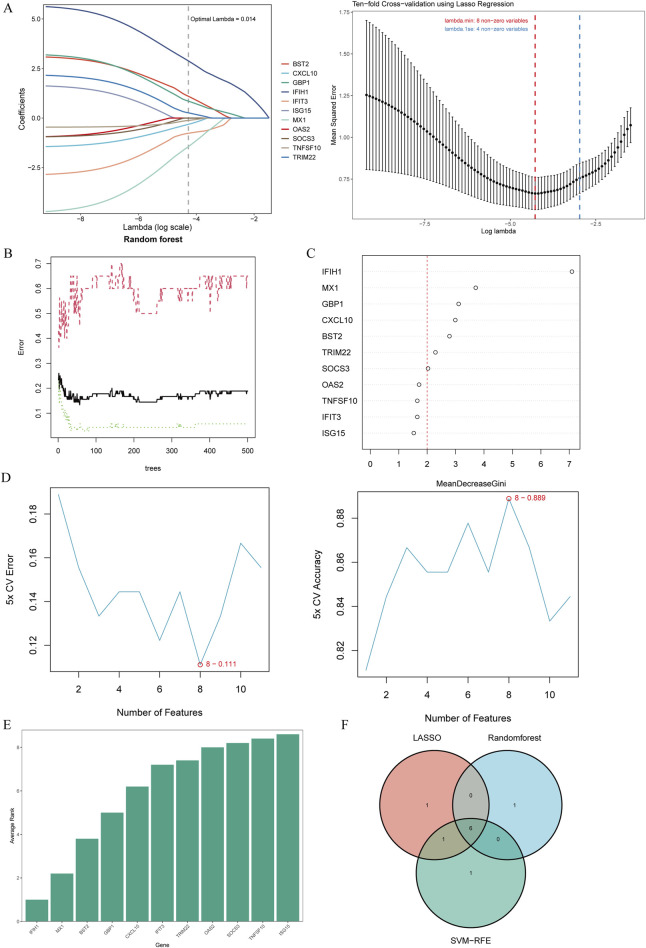
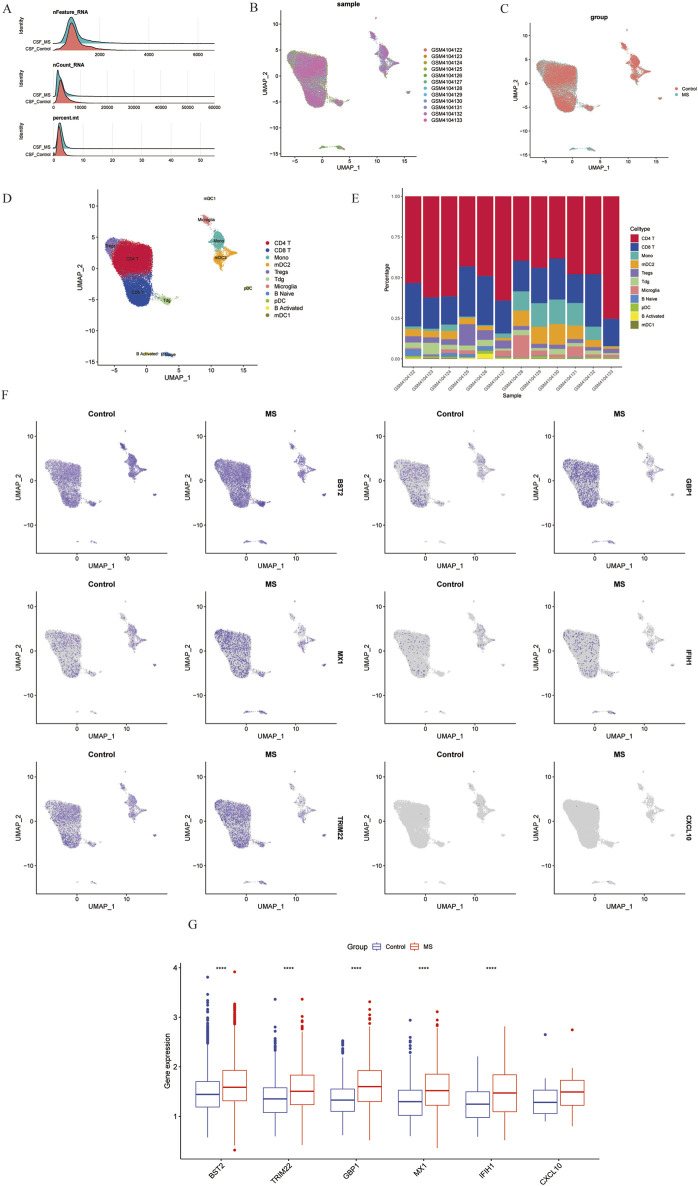
We retrieved transcriptomic data of MS, SLE, and RA patients from public databases. Weighted Gene Co-Expression Network Analysis (WGCNA) was employed to construct gene co-expression networks and identify disease-associated modules. Functional enrichment analyses and Protein-Protein Interaction (PPI) network was constructed. We used machine learning algorithms to select candidate biomarkers and evaluate their diagnostic value. The Cibersort algorithm was and scRNA-seq analysis was performed to identify key gene expression patterns and assess the infiltration of immune cells in MS patients. Finally, the biomarkers' expression was validated in human and mice experiments.
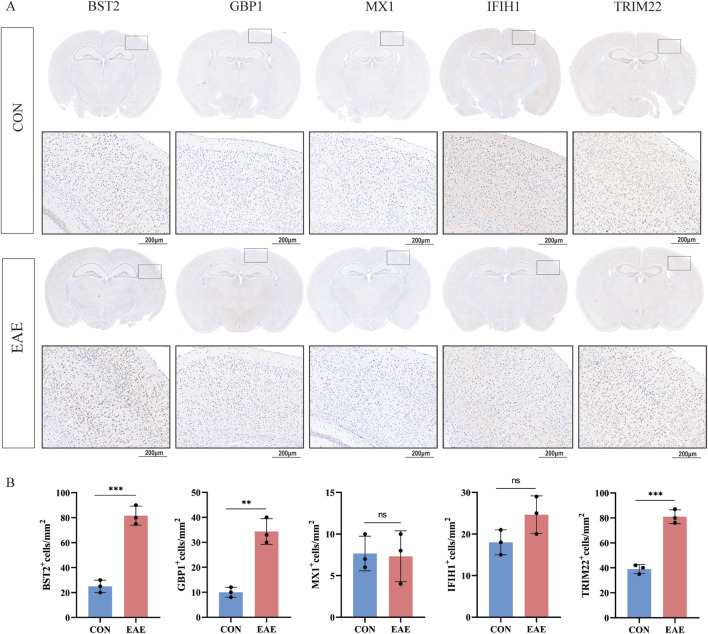
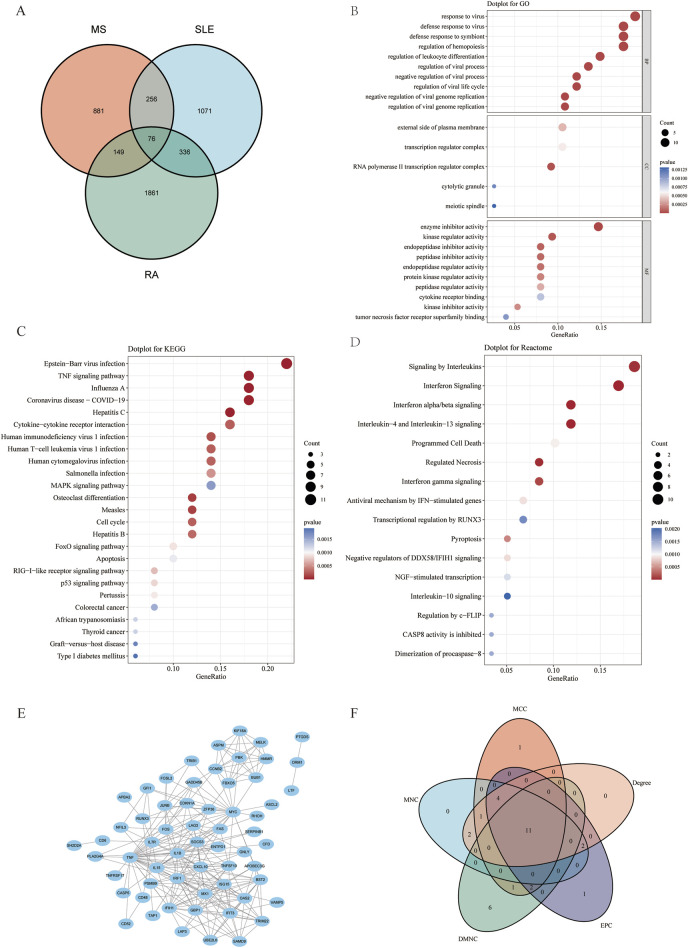
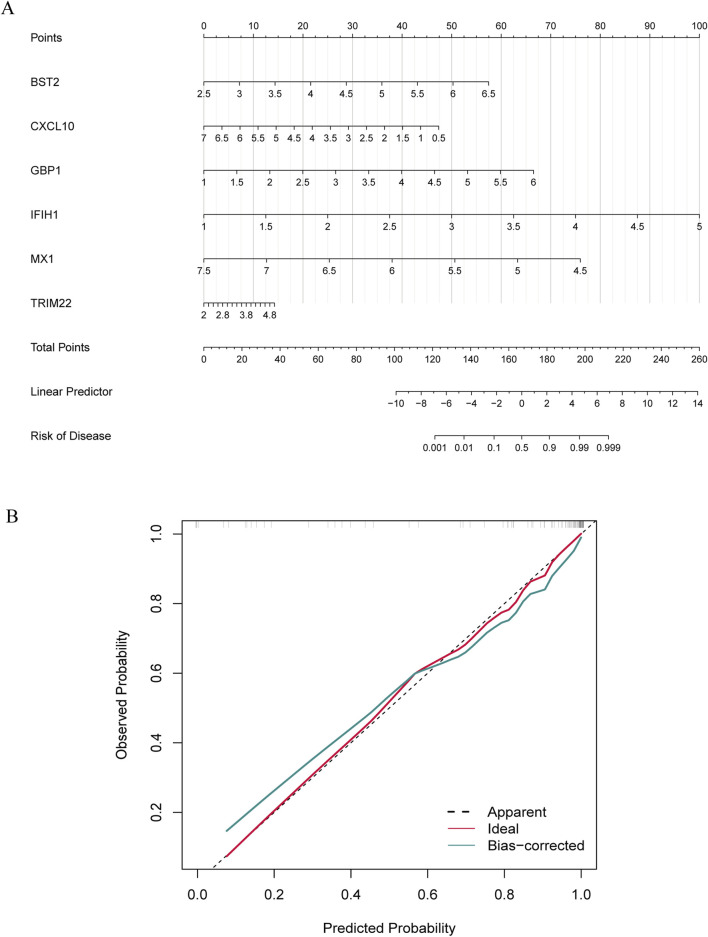
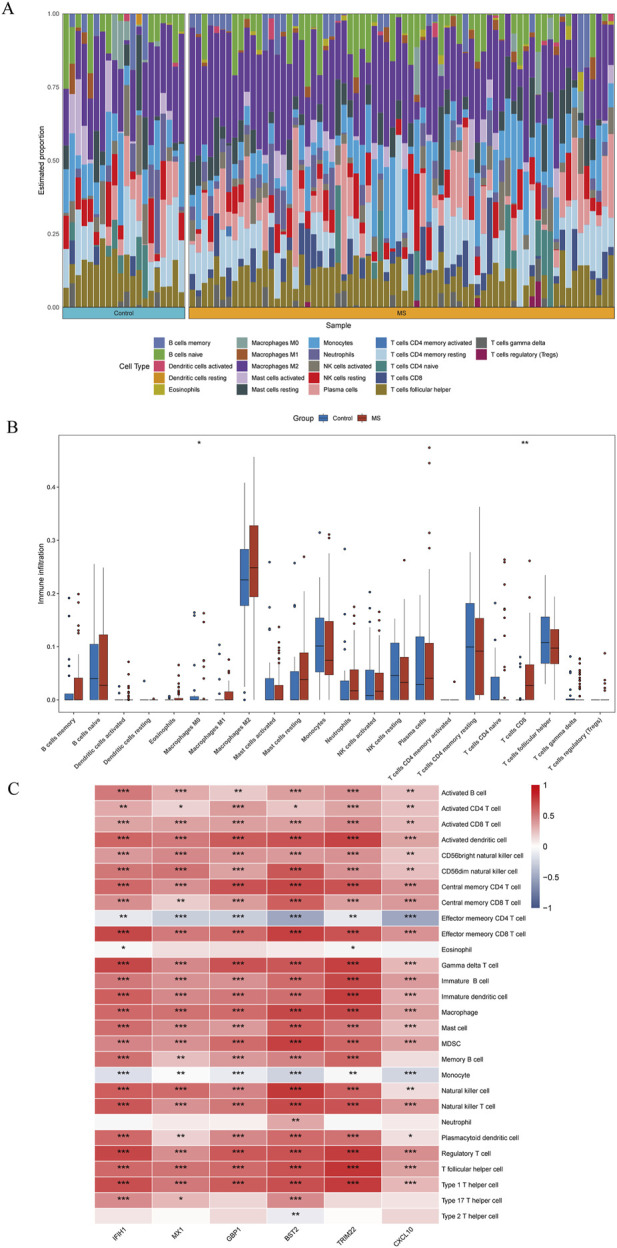
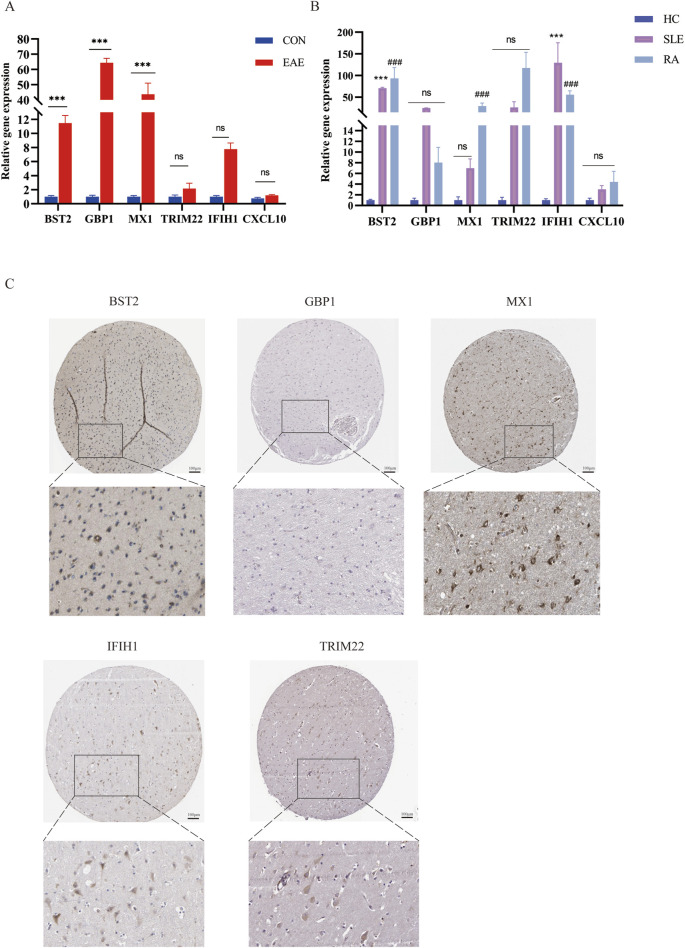
Several shared genes among MS, SLE, and RA were identified, which play crucial roles in immune responses and inflammation regulation. PPI network analysis highlighted key hub genes, some of which were selected as candidate biomarkers through machine learning algorithms. Receiver Operating Characteristic (ROC) curve analysis indicated that some genes had high diagnostic value (Area Under the Curve, AUC >0.7). Immune cell infiltration pattern analysis showed significant differences in the expression of various immune cells in MS patients. scRNA-seq analysis revealed clusters of genes that were significantly upregulated in the single cells of cerebrospinal fluid in MS patients. The expression of shared genes was validated in the EAE mose model. Validation using clinical samples confirmed the expression of potential diagnostic biomarkers.
This study identified shared genes among MS, SLE, and RA and proposed potential early diagnostic biomarkers. These genes are pivotal in regulating immune responses, providing new targets and theoretical basis for the early diagnosis and treatment of autoimmune diseases.
新出现的证据强调了自身免疫性疾病(AIDs)之间的共病机制,诸如单细胞RNA测序(scRNA-seq)等创新技术显著推动了该领域的探索。本研究旨在利用生物信息学数据库调查三种自身免疫性疾病——多发性硬化症(MS)、系统性红斑狼疮(SLE)和类风湿关节炎(RA)之间的共享基因,并确定早期诊断的潜在生物标志物。
我们从公共数据库中检索了MS、SLE和RA患者的转录组数据。采用加权基因共表达网络分析(WGCNA)构建基因共表达网络并识别疾病相关模块。进行功能富集分析并构建蛋白质-蛋白质相互作用(PPI)网络。我们使用机器学习算法选择候选生物标志物并评估其诊断价值。采用Cibersort算法并进行scRNA-seq分析,以识别关键基因表达模式并评估MS患者免疫细胞的浸润情况。最后,在人和小鼠实验中验证了生物标志物的表达。
确定了MS、SLE和RA之间的几个共享基因,它们在免疫反应和炎症调节中起关键作用。PPI网络分析突出了关键的枢纽基因,其中一些通过机器学习算法被选为候选生物标志物。受试者工作特征(ROC)曲线分析表明,一些基因具有较高的诊断价值(曲线下面积,AUC>0.7)。免疫细胞浸润模式分析显示,MS患者中各种免疫细胞的表达存在显著差异。scRNA-seq分析揭示了MS患者脑脊液单细胞中显著上调的基因簇。在实验性自身免疫性脑脊髓炎(EAE)小鼠模型中验证了共享基因的表达。使用临床样本进行的验证证实了潜在诊断生物标志物的表达。
本研究确定了MS、SLE和RA之间的共享基因,并提出了潜在的早期诊断生物标志物。这些基因在调节免疫反应中起关键作用,为自身免疫性疾病的早期诊断和治疗提供了新的靶点和理论基础。