Department of Biomolecular Medicine, Ghent University, Ghent, Belgium; Cancer Research Institute Ghent, Ghent, Belgium.
Department of Pediatrics, Baylor College of Medicine, Texas Children's Hospital Cancer Center, Houston, TX, USA.
Genome Biol. 2023 Aug 1;24(1):177. doi: 10.1186/s13059-023-03016-6.
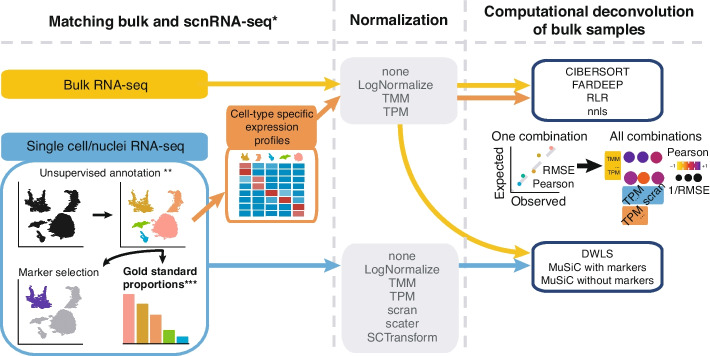
RNA profiling technologies at single-cell resolutions, including single-cell and single-nuclei RNA sequencing (scRNA-seq and snRNA-seq, scnRNA-seq for short), can help characterize the composition of tissues and reveal cells that influence key functions in both healthy and disease tissues. However, the use of these technologies is operationally challenging because of high costs and stringent sample-collection requirements. Computational deconvolution methods that infer the composition of bulk-profiled samples using scnRNA-seq-characterized cell types can broaden scnRNA-seq applications, but their effectiveness remains controversial.
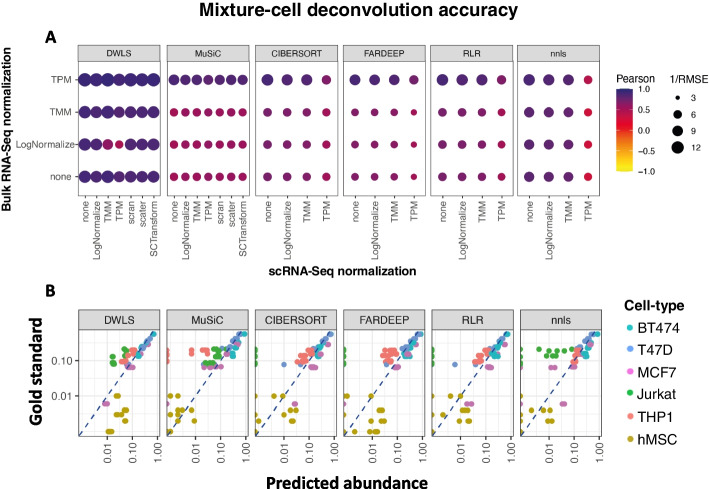
We produced the first systematic evaluation of deconvolution methods on datasets with either known or scnRNA-seq-estimated compositions. Our analyses revealed biases that are common to scnRNA-seq 10X Genomics assays and illustrated the importance of accurate and properly controlled data preprocessing and method selection and optimization. Moreover, our results suggested that concurrent RNA-seq and scnRNA-seq profiles can help improve the accuracy of both scnRNA-seq preprocessing and the deconvolution methods that employ them. Indeed, our proposed method, Single-cell RNA Quantity Informed Deconvolution (SQUID), which combines RNA-seq transformation and dampened weighted least-squares deconvolution approaches, consistently outperformed other methods in predicting the composition of cell mixtures and tissue samples.
We showed that analysis of concurrent RNA-seq and scnRNA-seq profiles with SQUID can produce accurate cell-type abundance estimates and that this accuracy improvement was necessary for identifying outcomes-predictive cancer cell subclones in pediatric acute myeloid leukemia and neuroblastoma datasets. These results suggest that deconvolution accuracy improvements are vital to enabling its applications in the life sciences.
单细胞分辨率的 RNA 分析技术,包括单细胞和单细胞核 RNA 测序(scRNA-seq 和 snRNA-seq,简称 scnRNA-seq),可以帮助描述组织的组成,并揭示对健康和疾病组织的关键功能有影响的细胞。然而,由于成本高和样本采集要求严格,这些技术的应用具有挑战性。使用 scnRNA-seq 鉴定的细胞类型来推断批量分析样本组成的计算去卷积方法可以拓宽 scnRNA-seq 的应用,但它们的有效性仍存在争议。
我们首次对具有已知或 scnRNA-seq 估计组成的数据集的去卷积方法进行了系统评估。我们的分析揭示了 scnRNA-seq 10X Genomics 测定中常见的偏差,并说明了准确和适当控制数据预处理以及方法选择和优化的重要性。此外,我们的结果表明,同时进行 RNA-seq 和 scnRNA-seq 分析可以帮助提高 scnRNA-seq 预处理和使用它们的去卷积方法的准确性。事实上,我们提出的方法,即单细胞 RNA 定量信息去卷积(SQUID),它结合了 RNA-seq 转换和衰减加权最小二乘去卷积方法,在预测细胞混合物和组织样本的组成方面始终优于其他方法。
我们表明,使用 SQUID 分析同时进行的 RNA-seq 和 scnRNA-seq 分析可以产生准确的细胞类型丰度估计,并且这种准确性的提高对于识别儿科急性髓系白血病和神经母细胞瘤数据集的具有预测结局的癌症细胞亚克隆是必要的。这些结果表明,去卷积准确性的提高对于使其在生命科学中的应用至关重要。