Afeke Innocent, Adu-Amankwaah Joseph, Ankrah Lennox Mac, Orish Verner Ndudiri, Jamfaru Ibrahim, Hamid Abdul-Wahab Mawuko, Amegan-Aho Kokou Hefoume, Mbroh Hintermann Kobina, Mensah Graceful Lord, Ablordey Anthony Samuel
Department of Medical Laboratory Sciences, School of Allied Health Sciences, University of Health and Allied Sciences, Ho, Ghana.
Department of Physiology, Xuzhou Medical University, Xuzhou, Jiangsu, China.
Pan Afr Med J. 2025 Feb 14;50:53. doi: 10.11604/pamj.2025.50.53.44554. eCollection 2025.
despite ongoing efforts, the health burden of neonatal infection remains unacceptably high due to major challenges, including the hurdle of determining the origin of infection. In this study, we explored the combination of tuf gene sequencing and the Maximum Likelihood Phylogenetic Model (MLPM) as a possible method for investigating the source(s) of transmission of two staphylococcal species, S. epidermidis and S. haemolyticus to neonates and young infants in the Ho Teaching Hospital (HTH) of Ghana, where we previously identified bloodstream infections as a major cause of neonatal morbidity and mortality.
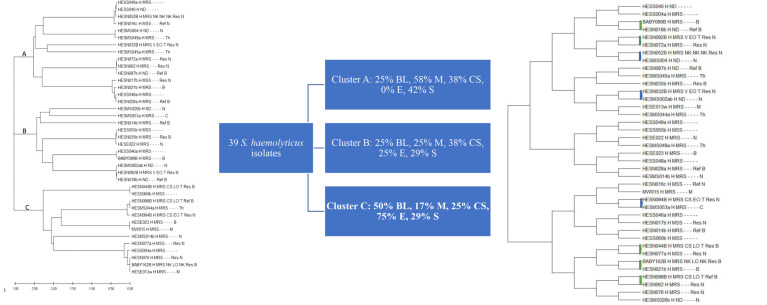
a total of 106 bacterial isolates were analyzed, comprising 67 S. epidermidis and 39 S. haemolyticus, cultured from blood samples of neonates and young infants, nasal mucosae of mothers, clinical staff, students, and a few objects in the hospital. Isolates were identified using Bruker Daltonik MALDI-TOF, and their nucleic acids were obtained. The tuf genes were sequenced using the Sanger method, and bioinformatics analyses were performed using the MEGA5 (10.1.8 version).
from our data, the combined use of bacterial tuf gene sequencing and MLPM revealed that mothers are the main source of S. haemolyticus-associated neonatal infections, whereas clinical staff is more likely to transmit S. epidermidis to neonates and young infants in the HTH. Whole genome sequencing scatter plots of a few of the isolates were used as a comparator method.
overall, our findings suggest that using bacterial tuf gene sequencing in conjunction with bioinformatic analysis of this gene utilizing the MLPM may serve as a useful epidemiologic method in predicting the source of staphylococcal infections in neonates in the Neonatal Intensive Care Unit (NICU) and possibly other units in the hospital.
尽管一直在努力,但由于存在重大挑战,包括确定感染源这一障碍,新生儿感染的健康负担仍然高得令人无法接受。在本研究中,我们探索了tuf基因测序与最大似然系统发育模型(MLPM)相结合的方法,作为调查加纳霍教学医院(HTH)新生儿和幼儿感染表皮葡萄球菌和溶血葡萄球菌这两种葡萄球菌的传播源的一种可能方法,我们之前在该医院确定血流感染是新生儿发病和死亡的主要原因。
共分析了106株细菌分离株,其中包括从新生儿和幼儿的血样、母亲的鼻粘膜、临床工作人员、学生以及医院的一些物品中培养出的67株表皮葡萄球菌和39株溶血葡萄球菌。使用布鲁克道尔顿公司的基质辅助激光解吸电离飞行时间质谱仪(Bruker Daltonik MALDI-TOF)鉴定分离株,并获取其核酸。采用桑格法对tuf基因进行测序,并使用MEGA5(10.1.8版本)进行生物信息学分析。
根据我们的数据,细菌tuf基因测序与MLPM的联合使用表明,母亲是与溶血葡萄球菌相关的新生儿感染的主要来源,而在HTH,临床工作人员更有可能将表皮葡萄球菌传播给新生儿和幼儿。一些分离株的全基因组测序散点图被用作比较方法。
总体而言,我们的研究结果表明,将细菌tuf基因测序与利用MLPM对该基因进行生物信息学分析相结合,可能是一种有用的流行病学方法,可用于预测新生儿重症监护病房(NICU)以及医院其他可能科室中新生儿葡萄球菌感染的来源。