Avraham-Davidi Inbal, Mages Simon, Klughammer Johanna, Moriel Noa, Imada Shinya, Hofree Matan, Murray Evan, Chen Jonathan, Pelka Karin, Mehta Arnav, Boland Genevieve M, Delorey Toni, Caplan Leah, Dionne Danielle, Strasser Robert, Lalakova Jana, Niesnerova Anezka, Xu Hao, Rouault Morgane, Tirosh Itay, Hacohen Nir, Chen Fei, Yilmaz Omer, Roper Jatin, Rozenblatt-Rosen Orit, Nitzan Mor, Regev Aviv
Klarman Cell Observatory, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
These authors contributed equally.
bioRxiv. 2025 Jun 24:2022.10.02.508492. doi: 10.1101/2022.10.02.508492.
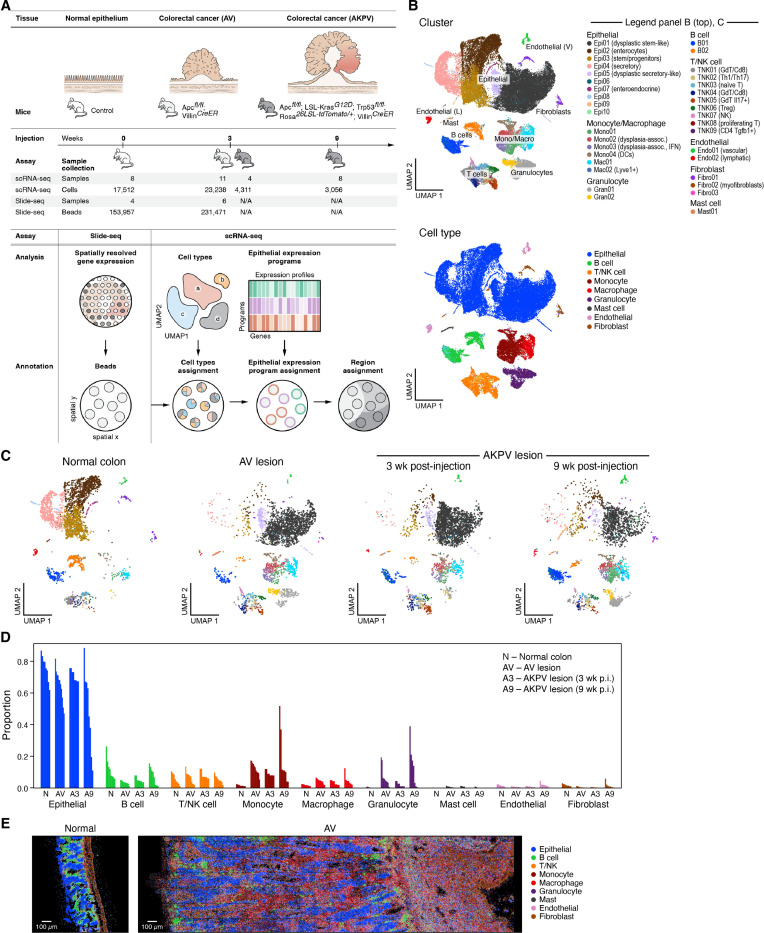
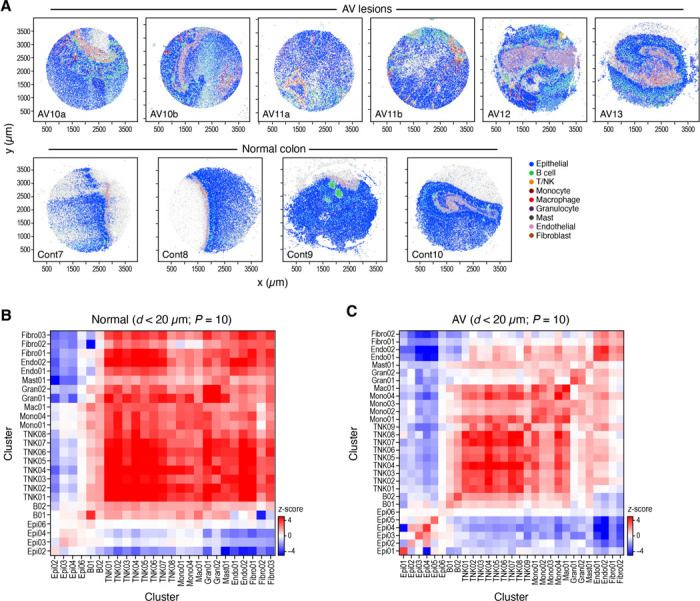
While advances in single cell genomics have helped to chart the cellular components of tumor ecosystems, it has been more challenging to characterize their specific spatial organization and functional interactions. Here, we combine single cell RNA-seq, spatial transcriptomics by Slide-seq, and multiplex RNA analysis, to create a detailed spatial map of healthy and dysplastic colon cellular ecosystems and their association with disease progression. We profiled inducible genetic CRC mouse models that recapitulate key features of human CRC, assigned cell types and epithelial expression programs to spatial tissue locations in tumors, and computationally used them to identify the regional features spanning different cells in the same spatial niche. We find that tumors were organized in cellular neighborhoods, each with a distinct composition of cell subtypes, expression programs, and local cellular interactions. Comparing to scRNA-seq and Slide-seq data from human CRC, we find that both cell composition and layout features were conserved between the species, with mouse neighborhoods correlating with malignancy and clinical outcome in human patient tumors, highlighting the relevance of our findings to human disease. Our work offers a comprehensive framework that is applicable across various tissues, tumors, and disease conditions, with tools for the extrapolation of findings from experimental mouse models to human diseases.
虽然单细胞基因组学的进展有助于描绘肿瘤生态系统的细胞组成,但表征其特定的空间组织和功能相互作用更具挑战性。在这里,我们结合单细胞RNA测序、通过Slide-seq进行的空间转录组学和多重RNA分析,以创建健康和发育异常的结肠细胞生态系统的详细空间图谱,以及它们与疾病进展的关联。我们对可诱导遗传的结直肠癌小鼠模型进行了分析,这些模型概括了人类结直肠癌的关键特征,将细胞类型和上皮表达程序分配到肿瘤中的空间组织位置,并通过计算利用它们来识别同一空间生态位中不同细胞的区域特征。我们发现肿瘤是由细胞邻域组成的,每个邻域都有不同的细胞亚型组成、表达程序和局部细胞相互作用。与来自人类结直肠癌的scRNA-seq和Slide-seq数据相比,我们发现物种之间的细胞组成和布局特征都是保守的,小鼠邻域与人类患者肿瘤的恶性程度和临床结果相关,突出了我们的发现与人类疾病的相关性。我们的工作提供了一个全面的框架,适用于各种组织、肿瘤和疾病状况,并提供了将实验小鼠模型的发现外推至人类疾病的工具。