Callejas-Hernández Francisco, Shiratori Mari, Sullivan Steven A, Blow Frances, Carlton Jane M
Department of Biology, Center for Genomics and Systems Biology, New York University, New York, New York, United States of America.
PLoS Pathog. 2025 Jul 23;21(7):e1013282. doi: 10.1371/journal.ppat.1013282. eCollection 2025 Jul.
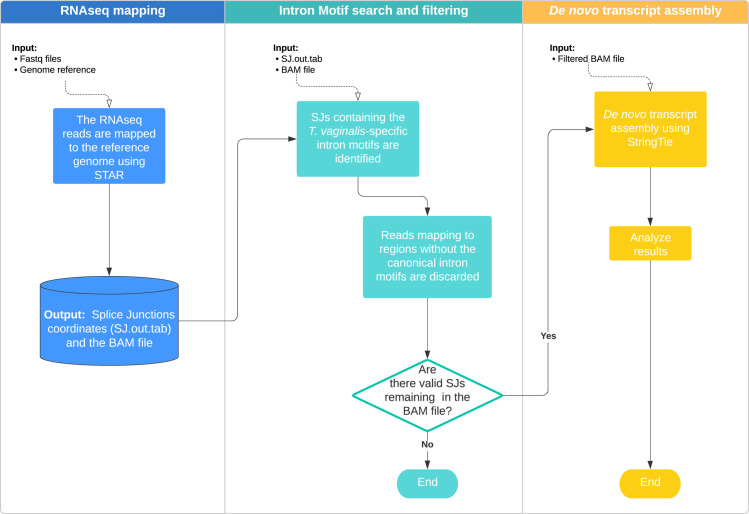
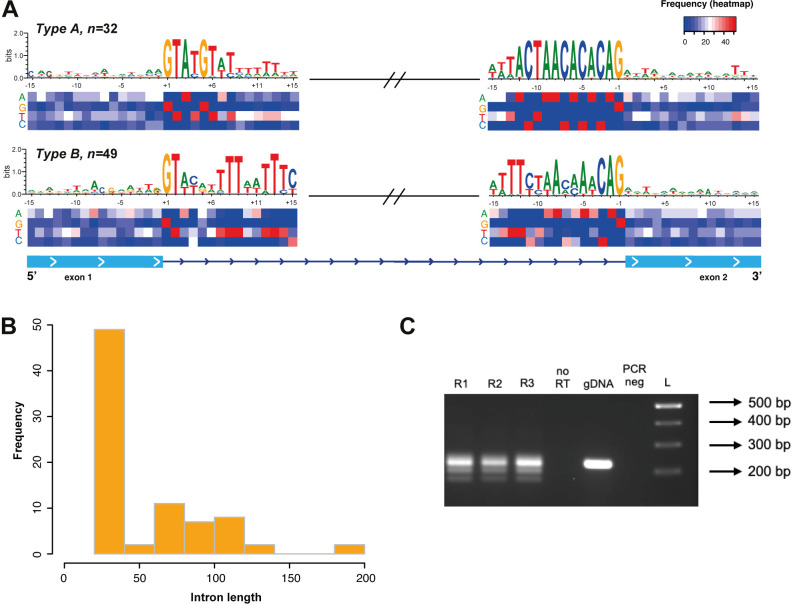
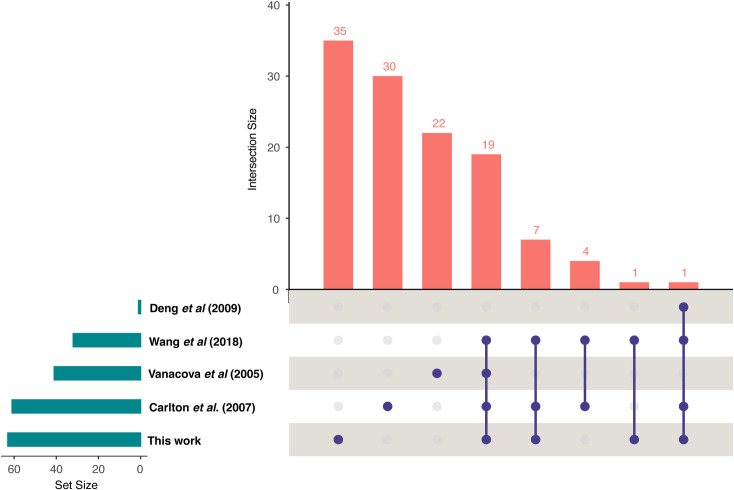
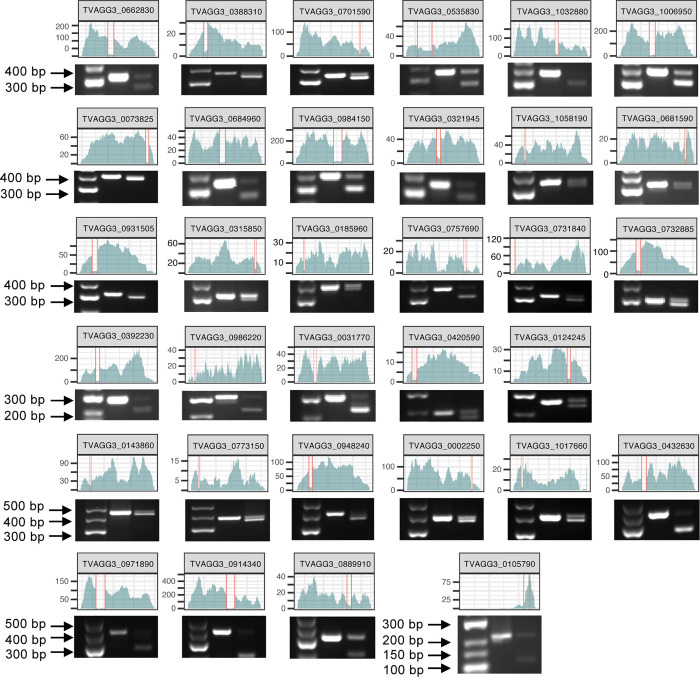
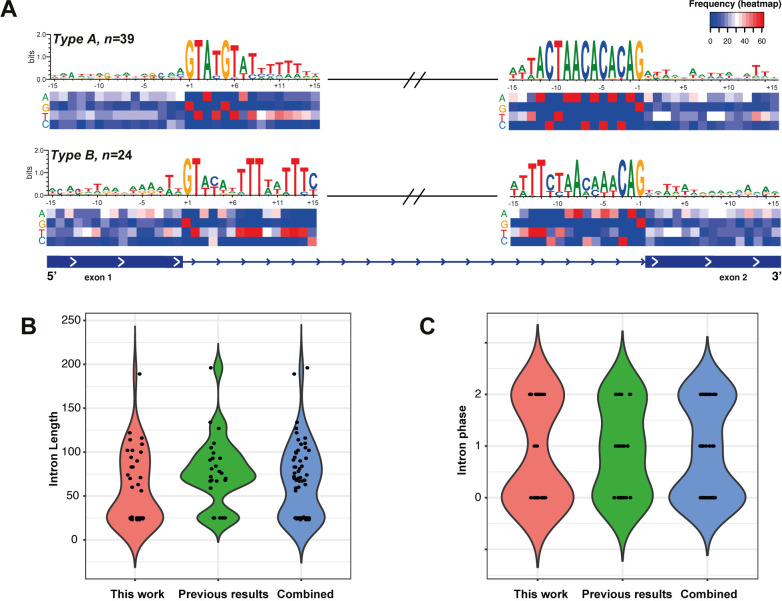
Trichomonas vaginalis infects the urogenital tract of men and women and causes the sexually transmitted infection trichomoniasis. Since the publication of its draft genome in 2007, the genome has drawn attention for several reasons, including its unusually large size, massive expansion of gene families, and high repeat content. The fragmented nature of the draft assembly made it challenging to obtain accurate metrics of features, such as spliceosomal introns. The number of introns identified has varied over the years, ranging from 41 when first characterized in 2005, to 32 in 2018 when the repertoire was revised. In both cases, the results suggested that more introns could be present in the genome. In this study, we exploited our new T. vaginalis G3 chromosome-scale assembly and annotation and high-coverage transcriptome datasets to provide an up-to-date repertoire of spliceosomal introns in the species. We developed a custom pipeline that distinguishes true splicing events from chimeric alignments by utilizing the extended motifs required by the splicing machinery, and experimentally verified the results using transcript evidence. We identified a total of 63 active introns and 34 putative "inactive" intron sequences in T. vaginalis, enabling an analysis of their length distribution, extended consensus motifs, intron phase distribution (including an unexpected expansion of UTR introns), and functional annotation. Notably, we found that a short intron in T. vaginalis, at only 23 nucleotides in size, is one of the shortest introns known to date. We tested our pipeline on a chromosome-scale assembly of the bird parasite Trichomonas stableri, the closest known relative to T. vaginalis. Our results revealed some conservation of the main features (total intron count, sequence, length distribution, and motifs) of these two closely related species, although differences in their functional annotation and duplication suggest alternative splicing machinery in T. vaginalis.
阴道毛滴虫感染男性和女性的泌尿生殖道,引起性传播感染滴虫病。自2007年其基因组草图公布以来,该基因组因多种原因受到关注,包括其异常大的尺寸、基因家族的大量扩张以及高重复含量。草图组装的碎片化性质使得获取诸如剪接体内含子等特征的准确指标具有挑战性。多年来鉴定出的内含子数量各不相同,从2005年首次表征时的41个,到2018年对其全部内含子进行修订时的32个。在这两种情况下,结果都表明基因组中可能存在更多内含子。在本研究中,我们利用新的阴道毛滴虫G3染色体规模的组装和注释以及高覆盖度转录组数据集,提供该物种剪接体内含子的最新信息。我们开发了一个定制流程,通过利用剪接机制所需的扩展基序,将真正的剪接事件与嵌合比对区分开来,并使用转录证据对结果进行实验验证。我们在阴道毛滴虫中总共鉴定出63个活性内含子和34个推定的“非活性”内含子序列,从而能够分析它们的长度分布、扩展共有基序、内含子相位分布(包括UTR内含子的意外扩张)以及功能注释。值得注意的是,我们发现阴道毛滴虫中一个仅23个核苷酸大小的短内含子是迄今为止已知的最短内含子之一。我们在鸟类寄生虫斯氏毛滴虫(阴道毛滴虫已知的最亲近的亲属)的染色体规模组装上测试了我们的流程。我们的结果揭示了这两个密切相关物种的一些主要特征(内含子总数、序列、长度分布和基序)的保守性,尽管它们在功能注释和重复方面的差异表明阴道毛滴虫中存在替代剪接机制。