Fu Yingqiang, Liu Yiyang, Sui Ziqi
Department of Breast Surgery, Second Affiliated Hospital, Zhejiang University School of Medicine, Linping Campus, Hangzhou City, 311100, Zhejiang Province, China.
Department of Gastroenterology, The Second Affiliated Hospital of Zhejiang Chinese Medical University, No. 318, Chaowang Road, Gongshu District, Hangzhou City, 310005, Zhejiang Province, China.
Discov Oncol. 2025 Jul 24;16(1):1400. doi: 10.1007/s12672-025-03084-z.
Per- and polyfluoroalkyl substances (PFAS), pervasive environmental contaminants, are increasingly linked to breast cancer, yet their molecular mechanisms remain unclear. This study integrates network toxicology and bioinformatics to elucidate PFAS-associated pathways and prognostic biomarkers in breast cancer.
Using the TCGA-BRCA dataset, we identified differentially expressed genes (DEGs) between normal and breast cancer tissues. We cross-referenced these genes with PFAS-related genes from the Comparative Toxicogenomics Database (CTD) to identify common targets. Enrichment analysis, network construction, and survival analysis were performed to elucidate the biological mechanisms and prognostic value. The CIBERSORT algorithm assessed immune cell infiltration, and molecular docking evaluated interactions between PFAS compounds and key genes.
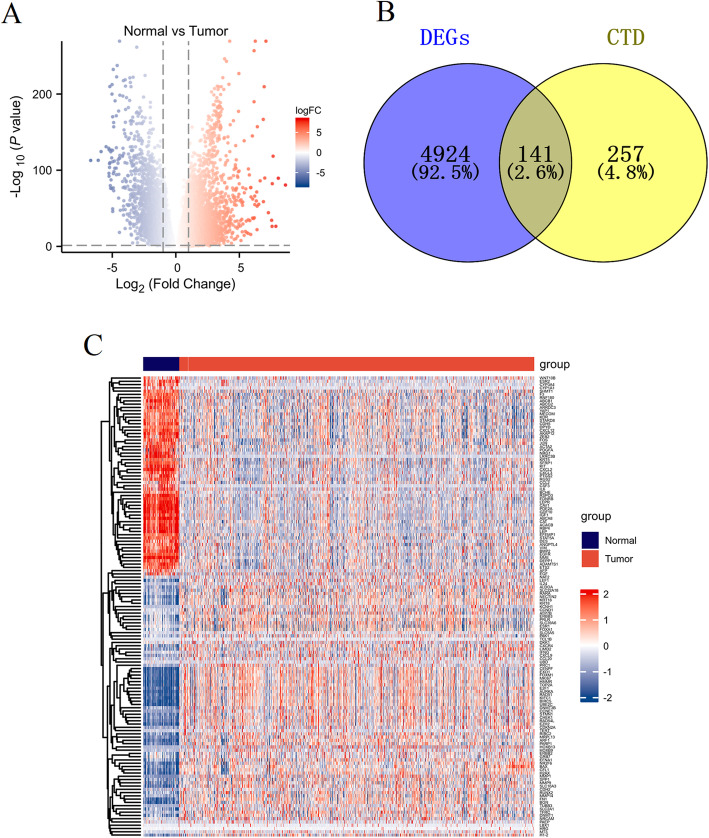
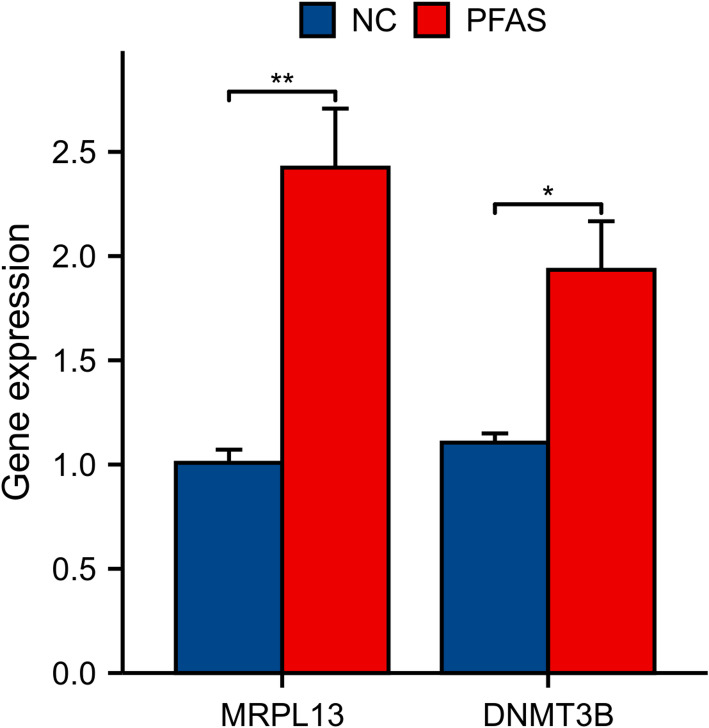
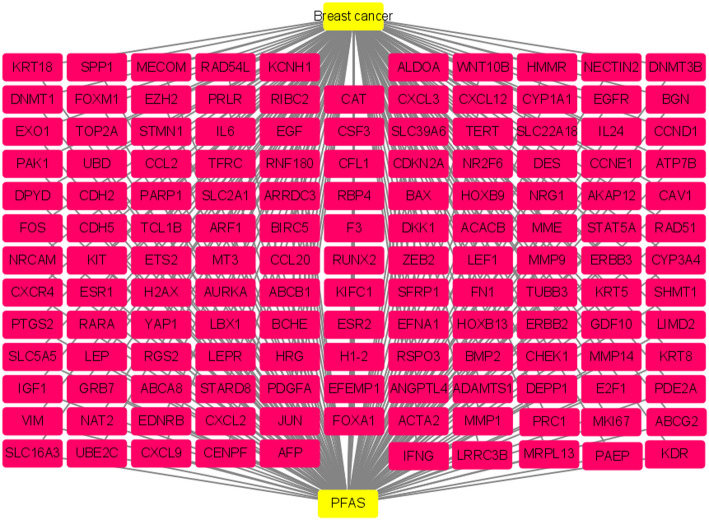
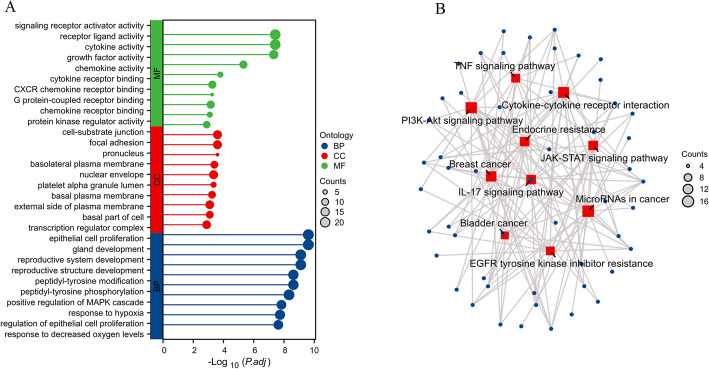
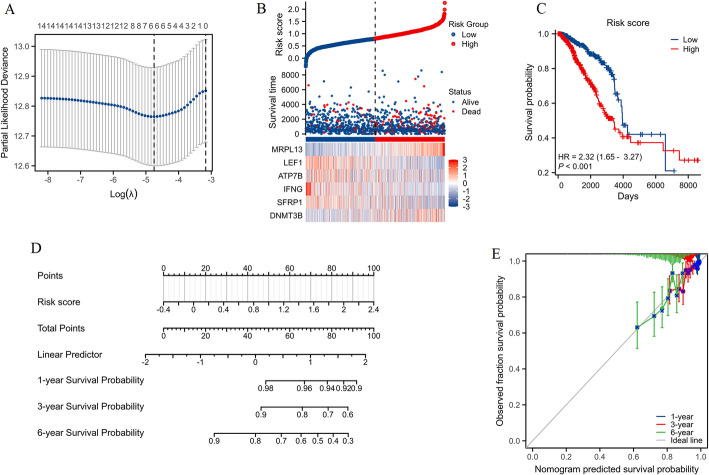
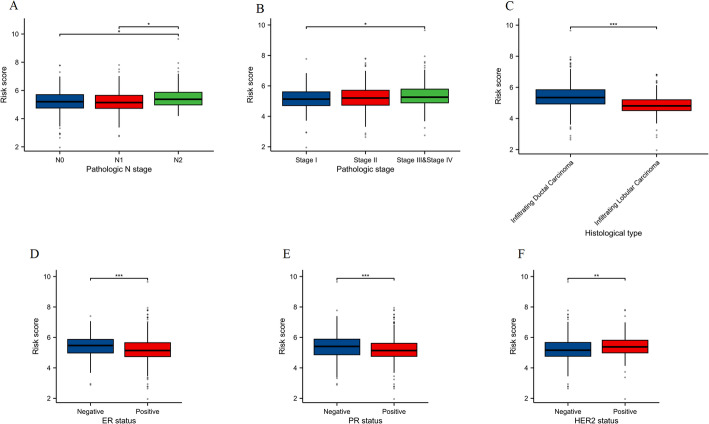
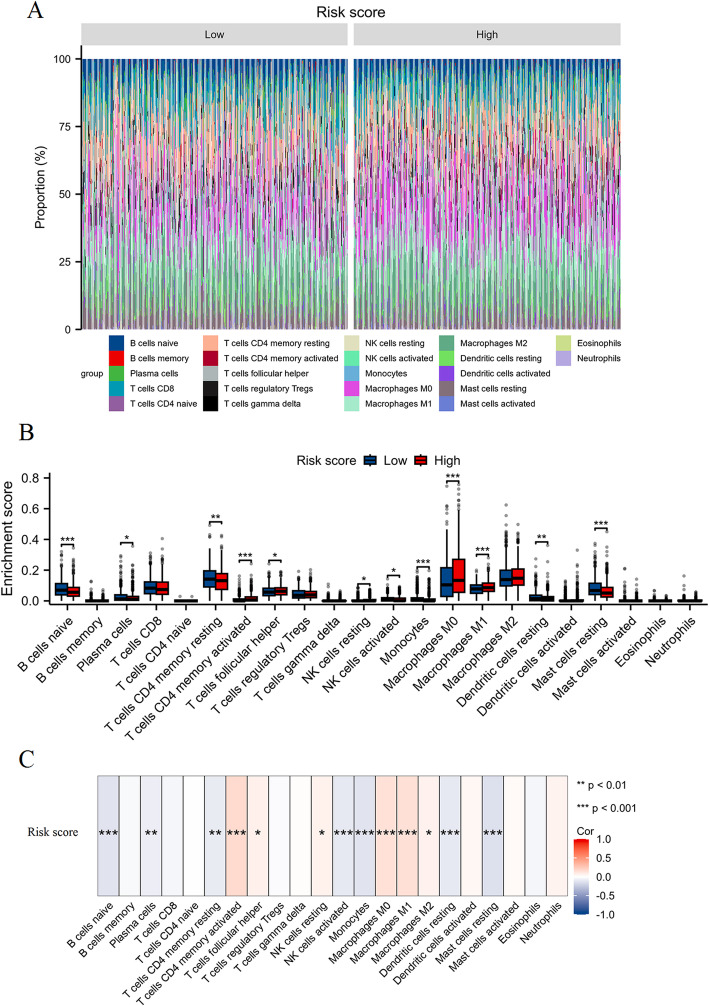
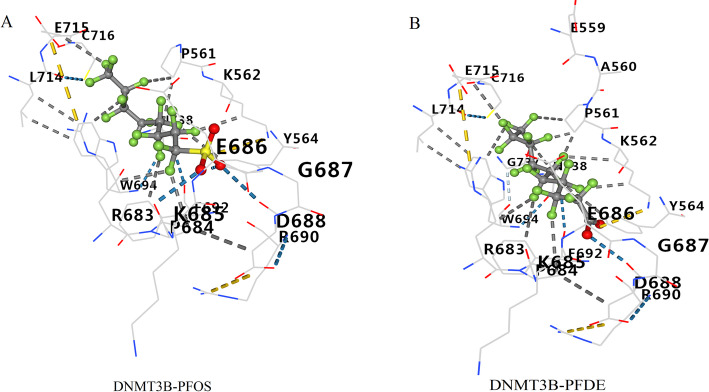
We identified 141 common DEGs, significantly enriched in pathways related to cytokine activity, growth factor activity, and chemokine receptor binding. A PFAS-toxicity target-breast cancer network illustrated potential mechanistic pathways. Six key prognostic genes (MRPL13, LEF1, ATP7B, IFNG, SFRP1, DNMT3B) were identified, forming a risk model that stratified patients with significant differences in survival. Higher risk scores were associated with advanced stages, specific histological types, and hormone receptor statuses. Immune cell infiltration analysis revealed distinct profiles between high and low-risk groups, with high-risk patients exhibiting elevated activated T cells and macrophages. Molecular docking showed strong interactions between PFAS compounds (PFOS and PFDE) and DNMT3B, suggesting potential gene function disruptions.
PFAS exposure is linked to altered gene expression, immune cell infiltration, and potential disruptions in key genes, contributing to breast cancer development and progression. These findings provide insights into potential therapeutic targets and underline the importance of addressing environmental factors in breast cancer management.
全氟和多氟烷基物质(PFAS)是普遍存在的环境污染物,与乳腺癌的关联日益增加,但其分子机制仍不清楚。本研究整合网络毒理学和生物信息学,以阐明乳腺癌中与PFAS相关的通路和预后生物标志物。
利用TCGA-BRCA数据集,我们鉴定了正常组织和乳腺癌组织之间的差异表达基因(DEG)。我们将这些基因与来自比较毒理基因组学数据库(CTD)的PFAS相关基因进行交叉引用,以确定共同靶点。进行富集分析、网络构建和生存分析,以阐明生物学机制和预后价值。CIBERSORT算法评估免疫细胞浸润,分子对接评估PFAS化合物与关键基因之间的相互作用。
我们鉴定出141个共同的DEG,显著富集于与细胞因子活性、生长因子活性和趋化因子受体结合相关的通路。一个PFAS-毒性靶点-乳腺癌网络阐明了潜在的机制通路。确定了六个关键的预后基因(MRPL13、LEF1、ATP7B、IFNG、SFRP1、DNMT3B),形成了一个风险模型,该模型对生存有显著差异的患者进行分层。较高的风险评分与晚期、特定组织学类型和激素受体状态相关。免疫细胞浸润分析显示高风险组和低风险组之间有明显差异,高风险患者表现出活化T细胞和巨噬细胞升高。分子对接显示PFAS化合物(PFOS和PFDE)与DNMT3B之间有强烈相互作用,表明可能存在基因功能破坏。
PFAS暴露与基因表达改变、免疫细胞浸润以及关键基因的潜在破坏有关,促进了乳腺癌的发生和发展。这些发现为潜在的治疗靶点提供了见解,并强调了在乳腺癌管理中解决环境因素的重要性。