Pyrc Krzysztof, Jebbink Maarten F, Berkhout Ben, van der Hoek Lia
Department of Human Retrovirology, University of Amsterdam, Meibergdreef 15, 1105 AZ, Amsterdam, The Netherlands.
Virol J. 2004 Nov 17;1:7. doi: 10.1186/1743-422X-1-7.
Two human coronaviruses are known since the 1960s: HCoV-229E and HCoV-OC43. SARS-CoV was discovered in the early spring of 2003, followed by the identification of HCoV-NL63, the fourth member of the coronaviridae family that infects humans. In this study, we describe the genome structure and the transcription strategy of HCoV-NL63 by experimental analysis of the viral subgenomic mRNAs.
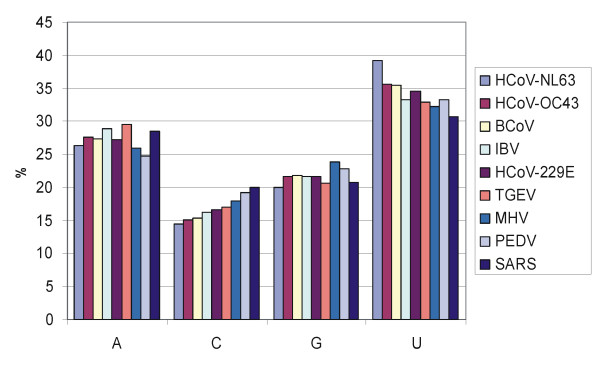
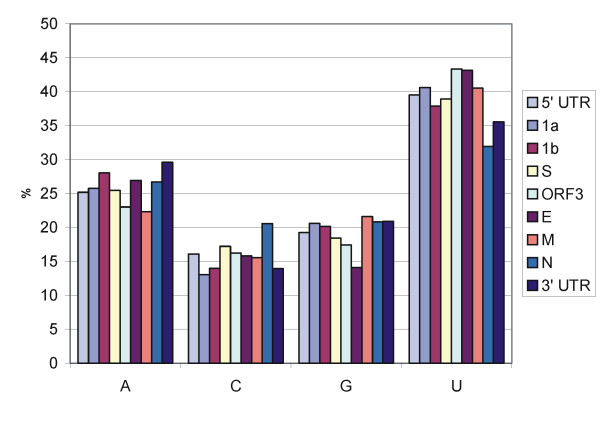
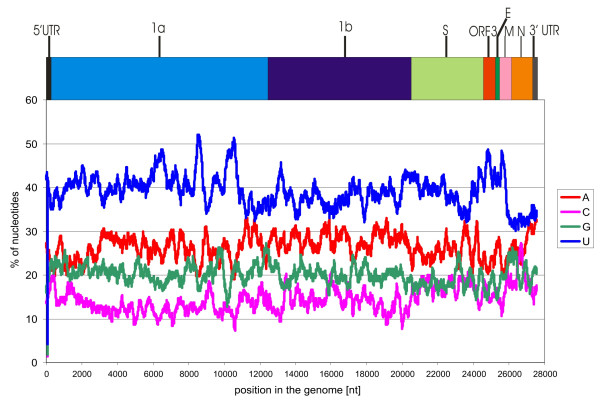
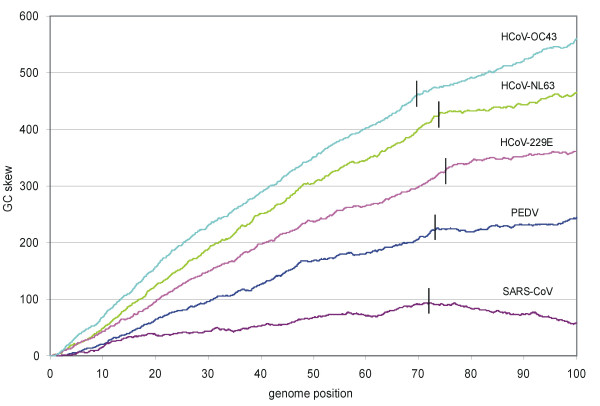
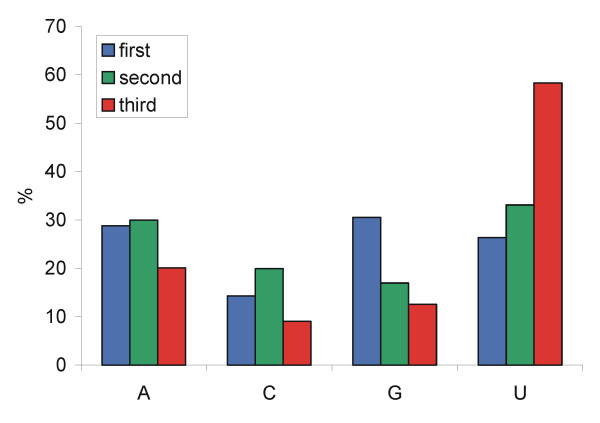
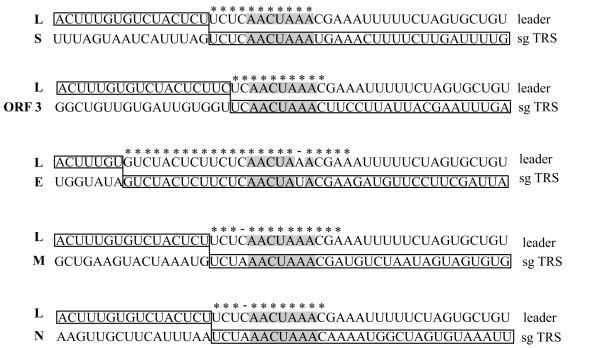
The genome of HCoV-NL63 has the following gene order: 1a-1b-S-ORF3-E-M-N. The GC content of the HCoV-NL63 genome is extremely low (34%) compared to other coronaviruses, and we therefore performed additional analysis of the nucleotide composition. Overall, the RNA genome is very low in C and high in U, and this is also reflected in the codon usage. Inspection of the nucleotide composition along the genome indicates that the C-count increases significantly in the last one-third of the genome at the expense of U and G. We document the production of subgenomic (sg) mRNAs coding for the S, ORF3, E, M and N proteins. We did not detect any additional sg mRNA. Furthermore, we sequenced the 5' end of all sg mRNAs, confirming the presence of an identical leader sequence in each sg mRNA. Northern blot analysis indicated that the expression level among the sg mRNAs differs significantly, with the sg mRNA encoding nucleocapsid (N) being the most abundant.
The presented data give insight into the viral evolution and mutational patterns in coronaviral genome. Furthermore our data show that HCoV-NL63 employs the discontinuous replication strategy with generation of subgenomic mRNAs during the (-) strand synthesis. Because HCoV-NL63 has a low pathogenicity and is able to grow easily in cell culture, this virus can be a powerful tool to study SARS coronavirus pathogenesis.
自20世纪60年代以来,已知两种人类冠状病毒:HCoV-229E和HCoV-OC43。严重急性呼吸综合征冠状病毒(SARS-CoV)于2003年早春被发现,随后鉴定出HCoV-NL63,它是冠状病毒科中感染人类的第四个成员。在本研究中,我们通过对病毒亚基因组mRNA的实验分析来描述HCoV-NL63的基因组结构和转录策略。
HCoV-NL63的基因组具有以下基因顺序:1a-1b-S-ORF3-E-M-N。与其他冠状病毒相比,HCoV-NL63基因组的鸟嘌呤-胞嘧啶(GC)含量极低(34%),因此我们对核苷酸组成进行了额外分析。总体而言,RNA基因组中的胞嘧啶(C)含量非常低,尿嘧啶(U)含量高,这也反映在密码子使用上。对基因组核苷酸组成的检查表明,在基因组的最后三分之一区域,C含量显著增加,而U和鸟嘌呤(G)含量减少。我们记录了编码刺突(S)、开放阅读框3(ORF3)、包膜(E)、膜(M)和核衣壳(N)蛋白的亚基因组(sg)mRNA的产生。我们未检测到任何其他sg mRNA。此外,我们对所有sg mRNA的5'端进行了测序,证实每个sg mRNA中都存在相同的前导序列。Northern印迹分析表明,sg mRNA之间的表达水平差异显著,编码核衣壳(N)的sg mRNA最为丰富。
所呈现的数据有助于深入了解冠状病毒基因组中的病毒进化和突变模式。此外,我们的数据表明,HCoV-NL63在负链合成过程中采用不连续复制策略产生亚基因组mRNA。由于HCoV-NL63致病性低且能够在细胞培养中轻松生长,这种病毒可成为研究SARS冠状病毒发病机制的有力工具。