Reiss David J, Baliga Nitin S, Bonneau Richard
Institute for Systems Biology, 1441 N, 34th St, Seattle, WA 98103-8904, USA.
BMC Bioinformatics. 2006 Jun 2;7:280. doi: 10.1186/1471-2105-7-280.
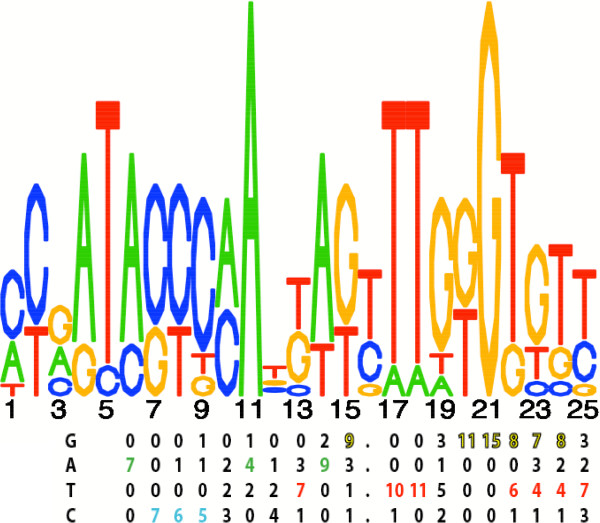
The learning of global genetic regulatory networks from expression data is a severely under-constrained problem that is aided by reducing the dimensionality of the search space by means of clustering genes into putatively co-regulated groups, as opposed to those that are simply co-expressed. Be cause genes may be co-regulated only across a subset of all observed experimental conditions, biclustering (clustering of genes and conditions) is more appropriate than standard clustering. Co-regulated genes are also often functionally (physically, spatially, genetically, and/or evolutionarily) associated, and such a priori known or pre-computed associations can provide support for appropriately grouping genes. One important association is the presence of one or more common cis-regulatory motifs. In organisms where these motifs are not known, their de novo detection, integrated into the clustering algorithm, can help to guide the process towards more biologically parsimonious solutions.
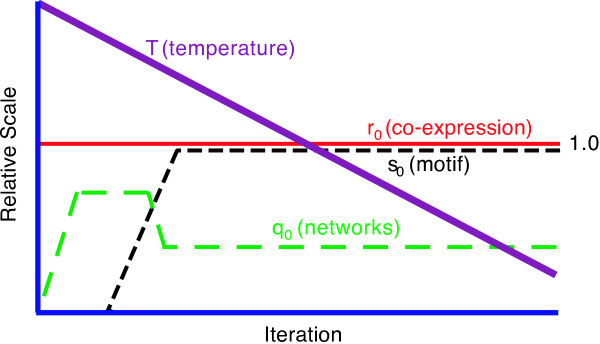
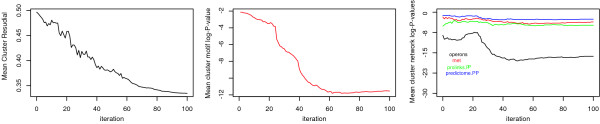
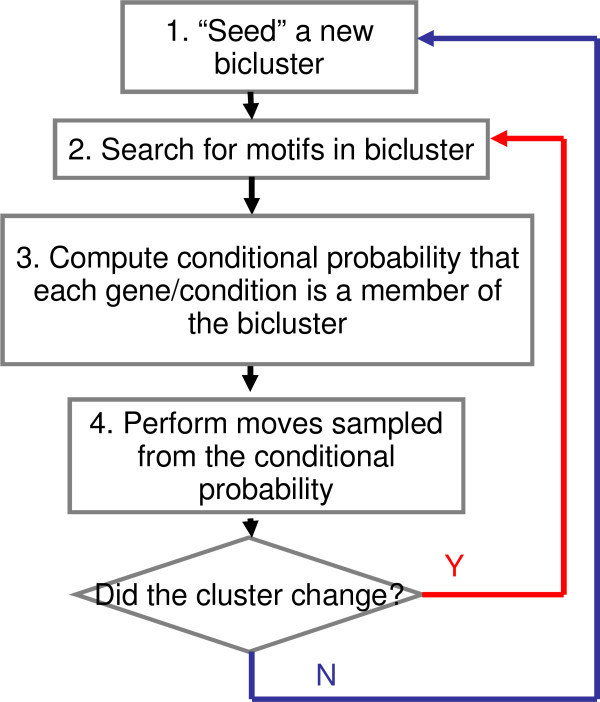
We have developed an algorithm, cMonkey, that detects putative co-regulated gene groupings by integrating the biclustering of gene expression data and various functional associations with the de novo detection of sequence motifs.
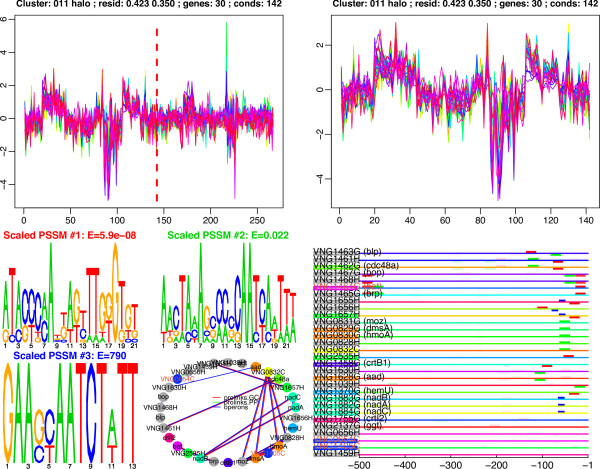
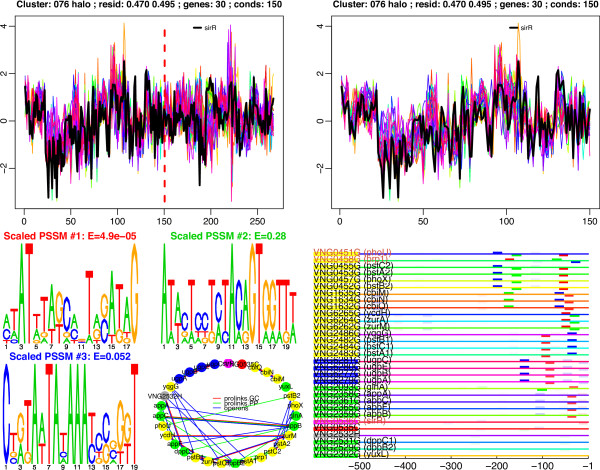
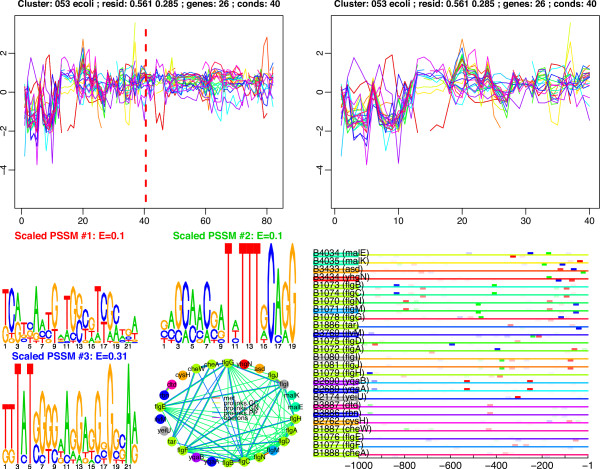
We have applied this procedure to the archaeon Halobacterium NRC-1, as part of our efforts to decipher its regulatory network. In addition, we used cMonkey on public data for three organisms in the other two domains of life: Helicobacter pylori, Saccharomyces cerevisiae, and Escherichia coli. The biclusters detected by cMonkey both recapitulated known biology and enabled novel predictions (some for Halobacterium were subsequently confirmed in the laboratory). For example, it identified the bacteriorhodopsin regulon, assigned additional genes to this regulon with apparently unrelated function, and detected its known promoter motif. We have performed a thorough comparison of cMonkey results against other clustering methods, and find that cMonkey biclusters are more parsimonious with all available evidence for co-regulation.
从表达数据中学习全局基因调控网络是一个严重缺乏约束的问题,通过将基因聚类到假定共同调控的组中(与仅仅是共同表达的组相对)来降低搜索空间的维度,有助于解决该问题。由于基因可能仅在所有观察到的实验条件的一个子集中受到共同调控,因此双聚类(基因和条件的聚类)比标准聚类更合适。共同调控的基因通常在功能上(物理、空间、遗传和/或进化方面)也相互关联,这种先验已知或预先计算的关联可以为适当地对基因进行分组提供支持。一个重要的关联是存在一个或多个共同的顺式调控基序。在这些基序未知的生物体中,将其从头检测整合到聚类算法中,可以帮助引导过程朝着更符合生物学简约性的解决方案发展。
我们开发了一种算法cMonkey,它通过整合基因表达数据的双聚类、各种功能关联以及序列基序的从头检测来检测假定的共同调控基因分组。
作为我们破译古菌嗜盐菌NRC - 1调控网络努力的一部分,我们将此程序应用于该古菌。此外,我们还将cMonkey应用于生命其他两个域中三种生物的公共数据:幽门螺杆菌、酿酒酵母和大肠杆菌。cMonkey检测到的双聚类既概括了已知生物学知识,又实现了新的预测(一些关于嗜盐菌的预测随后在实验室中得到证实)。例如,它识别出细菌视紫红质操纵子,为该操纵子分配了功能明显不相关的其他基因,并检测到其已知的启动子基序。我们将cMonkey的结果与其他聚类方法进行了全面比较,发现cMonkey双聚类在所有可用的共同调控证据方面更为简约。