Cho Young-Rae, Hwang Woochang, Ramanathan Murali, Zhang Aidong
Department of Computer Science and Engineering, State University of New York, Buffalo, NY, USA.
BMC Bioinformatics. 2007 Jul 24;8:265. doi: 10.1186/1471-2105-8-265.
The systematic analysis of protein-protein interactions can enable a better understanding of cellular organization, processes and functions. Functional modules can be identified from the protein interaction networks derived from experimental data sets. However, these analyses are challenging because of the presence of unreliable interactions and the complex connectivity of the network. The integration of protein-protein interactions with the data from other sources can be leveraged for improving the effectiveness of functional module detection algorithms.
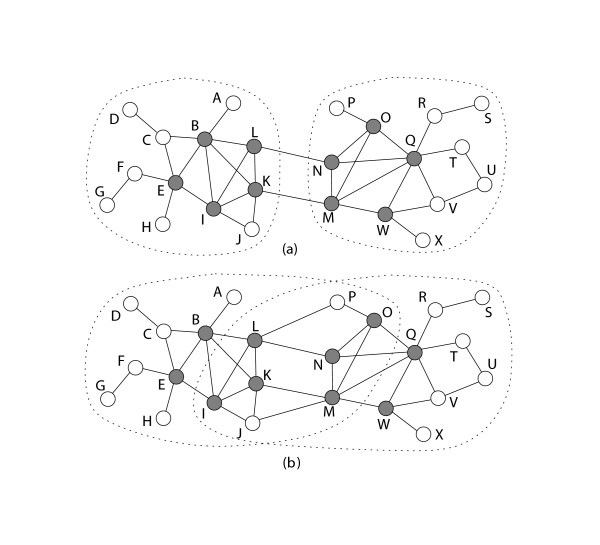
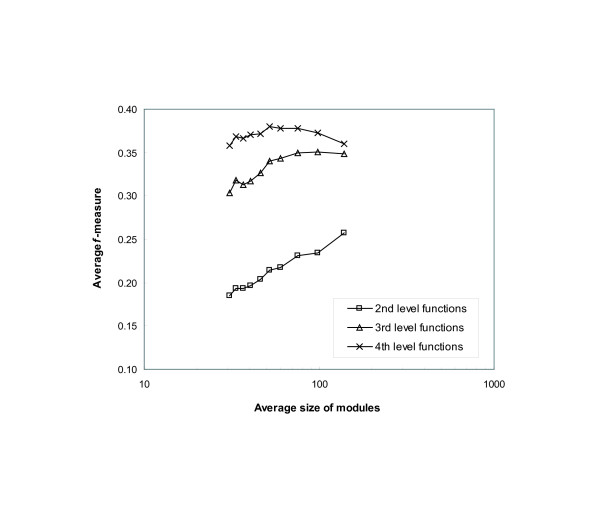
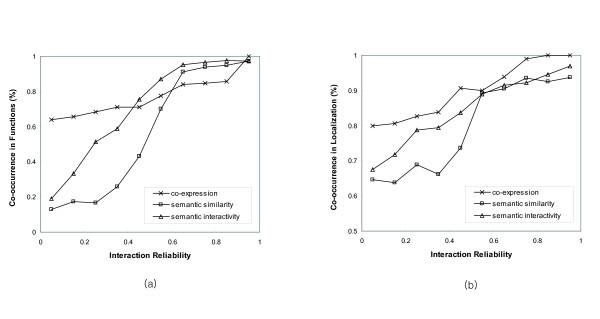
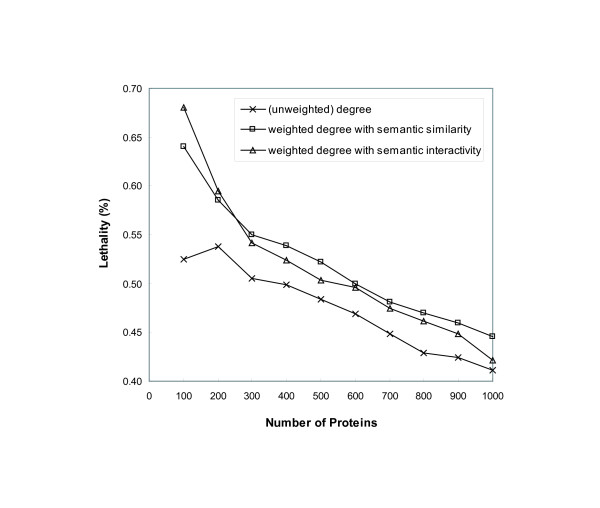
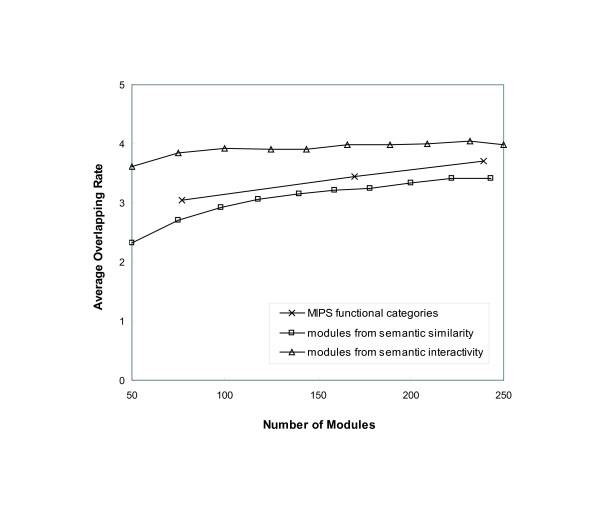
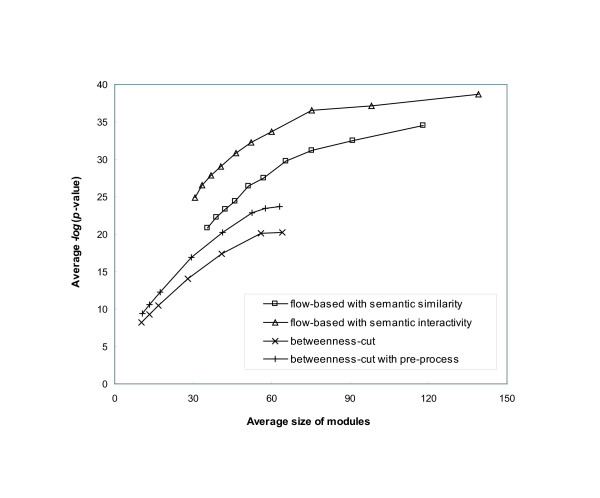
We have developed novel metrics, called semantic similarity and semantic interactivity, which use Gene Ontology (GO) annotations to measure the reliability of protein-protein interactions. The protein interaction networks can be converted into a weighted graph representation by assigning the reliability values to each interaction as a weight. We presented a flow-based modularization algorithm to efficiently identify overlapping modules in the weighted interaction networks. The experimental results show that the semantic similarity and semantic interactivity of interacting pairs were positively correlated with functional co-occurrence. The effectiveness of the algorithm for identifying modules was evaluated using functional categories from the MIPS database. We demonstrated that our algorithm had higher accuracy compared to other competing approaches.
The integration of protein interaction networks with GO annotation data and the capability of detecting overlapping modules substantially improve the accuracy of module identification.
对蛋白质-蛋白质相互作用进行系统分析有助于更好地理解细胞组织、过程和功能。可以从源自实验数据集的蛋白质相互作用网络中识别功能模块。然而,由于存在不可靠的相互作用以及网络的复杂连通性,这些分析具有挑战性。蛋白质-蛋白质相互作用与来自其他来源的数据的整合可用于提高功能模块检测算法的有效性。
我们开发了名为语义相似性和语义交互性的新指标,它们使用基因本体(GO)注释来衡量蛋白质-蛋白质相互作用的可靠性。通过将可靠性值作为权重分配给每个相互作用,可以将蛋白质相互作用网络转换为加权图表示。我们提出了一种基于流的模块化算法,以有效地识别加权相互作用网络中的重叠模块。实验结果表明,相互作用对的语义相似性和语义交互性与功能共现呈正相关。使用MIPS数据库中的功能类别评估了该算法识别模块的有效性。我们证明,与其他竞争方法相比,我们的算法具有更高的准确性。
蛋白质相互作用网络与GO注释数据的整合以及检测重叠模块的能力大大提高了模块识别的准确性。