Cokus Shawn J, Feng Suhua, Zhang Xiaoyu, Chen Zugen, Merriman Barry, Haudenschild Christian D, Pradhan Sriharsa, Nelson Stanley F, Pellegrini Matteo, Jacobsen Steven E
Department of Molecular, Cell, and Developmental Biology, University of California at Los Angeles, Los Angeles, California 90095, USA.
Nature. 2008 Mar 13;452(7184):215-9. doi: 10.1038/nature06745. Epub 2008 Feb 17.
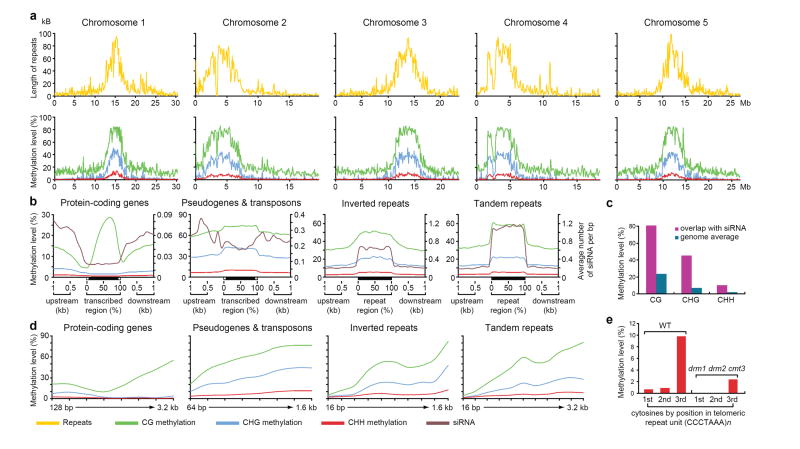
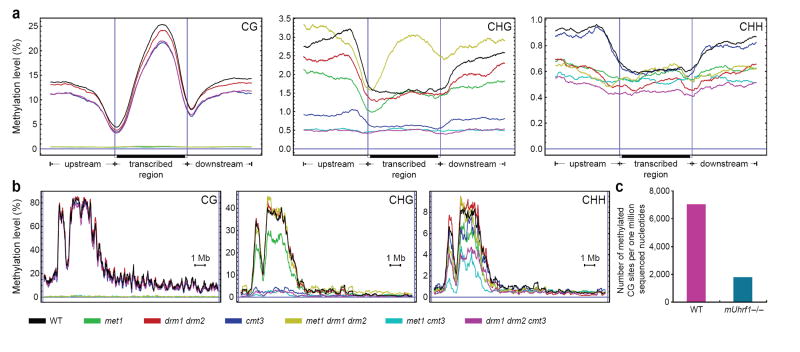
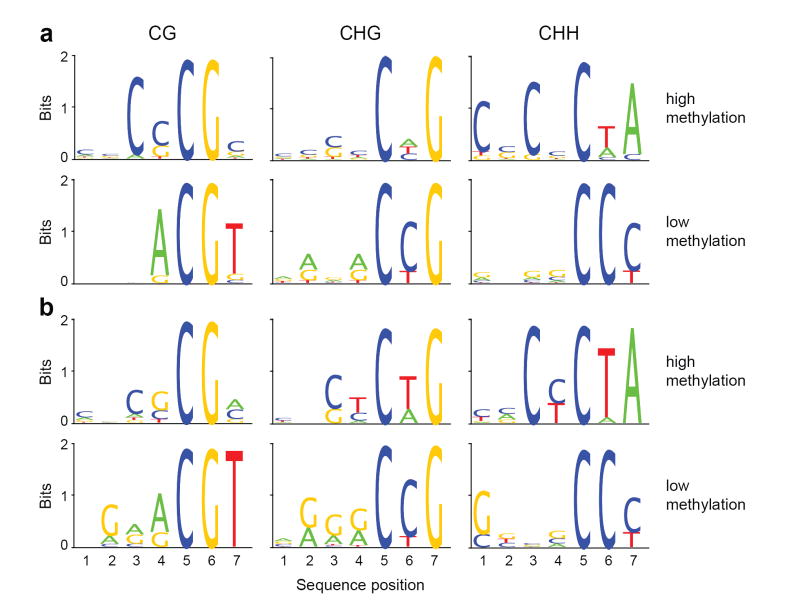
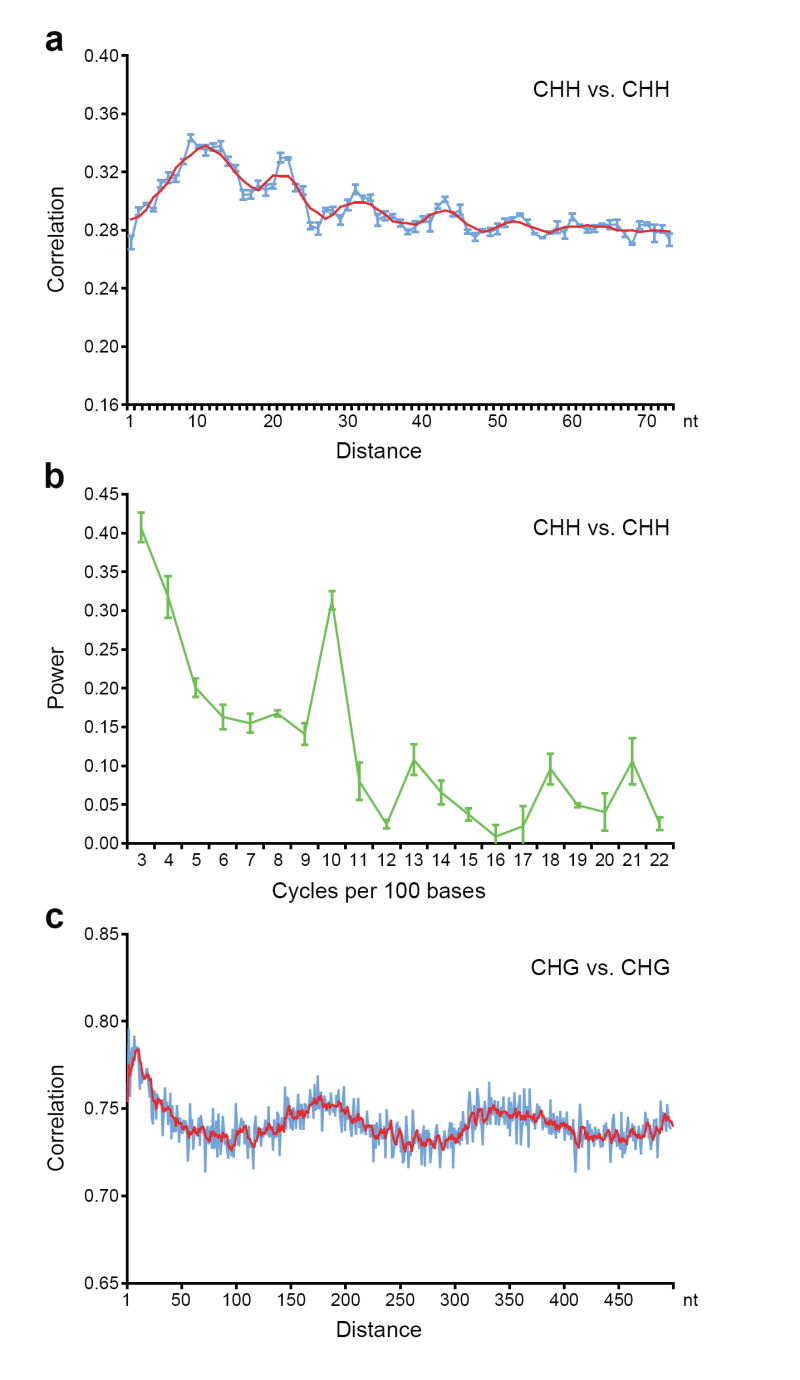
Cytosine DNA methylation is important in regulating gene expression and in silencing transposons and other repetitive sequences. Recent genomic studies in Arabidopsis thaliana have revealed that many endogenous genes are methylated either within their promoters or within their transcribed regions, and that gene methylation is highly correlated with transcription levels. However, plants have different types of methylation controlled by different genetic pathways, and detailed information on the methylation status of each cytosine in any given genome is lacking. To this end, we generated a map at single-base-pair resolution of methylated cytosines for Arabidopsis, by combining bisulphite treatment of genomic DNA with ultra-high-throughput sequencing using the Illumina 1G Genome Analyser and Solexa sequencing technology. This approach, termed BS-Seq, unlike previous microarray-based methods, allows one to sensitively measure cytosine methylation on a genome-wide scale within specific sequence contexts. Here we describe methylation on previously inaccessible components of the genome and analyse the DNA methylation sequence composition and distribution. We also describe the effect of various DNA methylation mutants on genome-wide methylation patterns, and demonstrate that our newly developed library construction and computational methods can be applied to large genomes such as that of mouse.
胞嘧啶DNA甲基化在调控基因表达以及沉默转座子和其他重复序列方面具有重要作用。近期对拟南芥的基因组研究表明,许多内源基因在其启动子区域或转录区域内发生甲基化,并且基因甲基化与转录水平高度相关。然而,植物具有由不同遗传途径控制的不同类型甲基化,目前缺乏关于任何给定基因组中每个胞嘧啶甲基化状态的详细信息。为此,我们通过将基因组DNA的亚硫酸氢盐处理与使用Illumina 1G基因组分析仪和Solexa测序技术的超高通量测序相结合,生成了拟南芥甲基化胞嘧啶的单碱基对分辨率图谱。这种方法称为BS-Seq,与以前基于微阵列的方法不同,它能够在特定序列背景下全基因组范围内灵敏地检测胞嘧啶甲基化。在此,我们描述了基因组中以前难以检测的成分上的甲基化情况,并分析了DNA甲基化序列的组成和分布。我们还描述了各种DNA甲基化突变体对全基因组甲基化模式的影响,并证明我们新开发的文库构建和计算方法可应用于如小鼠这样的大型基因组。