Kapustin Yuri, Souvorov Alexander, Tatusova Tatiana, Lipman David
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20814, USA.
Biol Direct. 2008 May 21;3:20. doi: 10.1186/1745-6150-3-20.
The computation of accurate alignments of cDNA sequences against a genome is at the foundation of modern genome annotation pipelines. Several factors such as presence of paralogs, small exons, non-consensus splice signals, sequencing errors and polymorphic sites pose recognized difficulties to existing spliced alignment algorithms.
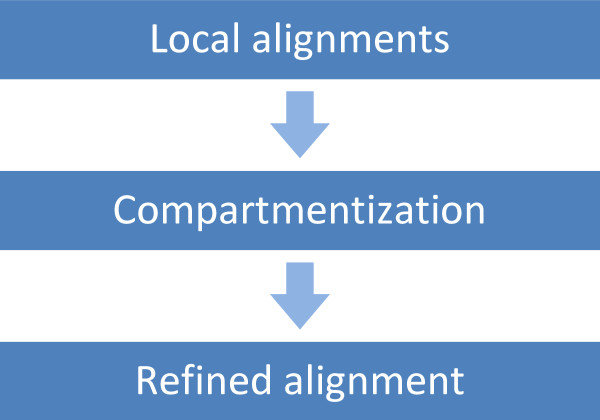
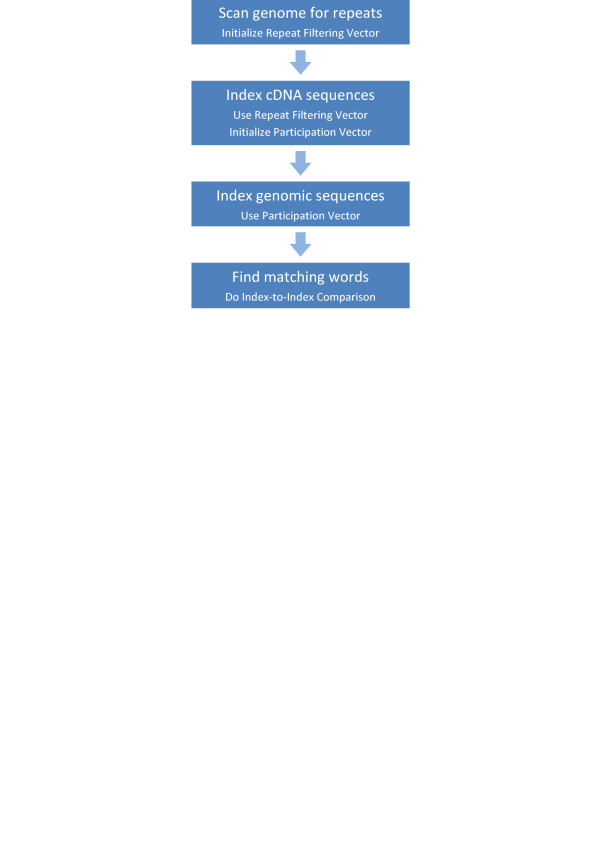
We describe a set of algorithms behind a tool called Splign for computing cDNA-to-Genome alignments. The algorithms include a high-performance preliminary alignment, a compartment identification based on a formally defined model of adjacent duplicated regions, and a refined sequence alignment. In a series of tests, Splign has produced more accurate results than other tools commonly used to compute spliced alignments, in a reasonable amount of time.
Splign's ability to deal with various issues complicating the spliced alignment problem makes it a helpful tool in eukaryotic genome annotation processes and alternative splicing studies. Its performance is enough to align the largest currently available pools of cDNA data such as the human EST set on a moderate-sized computing cluster in a matter of hours. The duplications identification (compartmentization) algorithm can be used independently in other areas such as the study of pseudogenes.
将cDNA序列与基因组进行精确比对的计算是现代基因组注释流程的基础。诸如旁系同源物的存在、小外显子、非一致性剪接信号、测序错误和多态性位点等多种因素给现有的剪接比对算法带来了公认的困难。
我们描述了一种名为Splign的工具背后的一组算法,用于计算cDNA到基因组的比对。这些算法包括高性能的初步比对、基于正式定义的相邻重复区域模型的区间识别以及精细的序列比对。在一系列测试中,Splign在合理的时间内产生了比其他常用于计算剪接比对的工具更准确的结果。
Splign处理使剪接比对问题复杂化的各种问题的能力使其成为真核生物基因组注释过程和可变剪接研究中的有用工具。它的性能足以在几小时内在中等规模的计算集群上对目前可用的最大cDNA数据集(如人类EST集)进行比对。重复序列识别(区间划分)算法可独立用于其他领域,如假基因研究。