Rogers Christopher S, Stoltz David A, Meyerholz David K, Ostedgaard Lynda S, Rokhlina Tatiana, Taft Peter J, Rogan Mark P, Pezzulo Alejandro A, Karp Philip H, Itani Omar A, Kabel Amanda C, Wohlford-Lenane Christine L, Davis Greg J, Hanfland Robert A, Smith Tony L, Samuel Melissa, Wax David, Murphy Clifton N, Rieke August, Whitworth Kristin, Uc Aliye, Starner Timothy D, Brogden Kim A, Shilyansky Joel, McCray Paul B, Zabner Joseph, Prather Randall S, Welsh Michael J
Department of Internal Medicine, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, IA 52242, USA.
Science. 2008 Sep 26;321(5897):1837-41. doi: 10.1126/science.1163600.
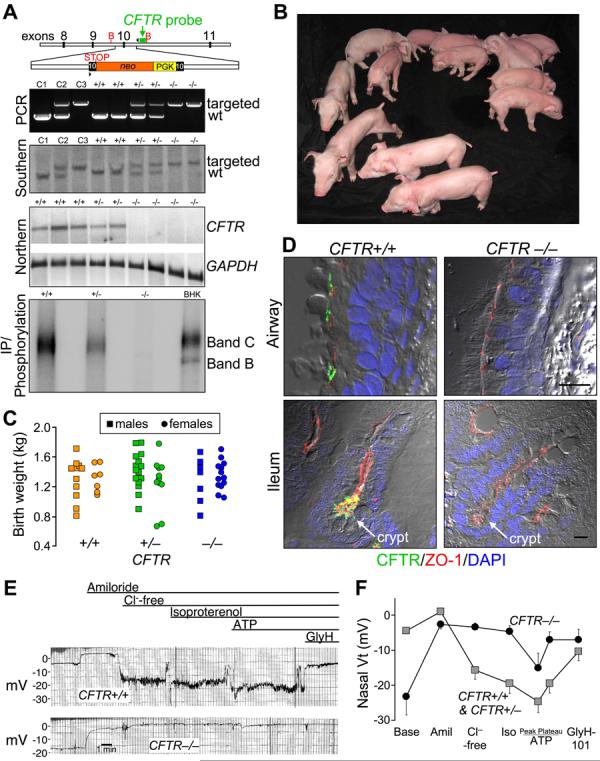
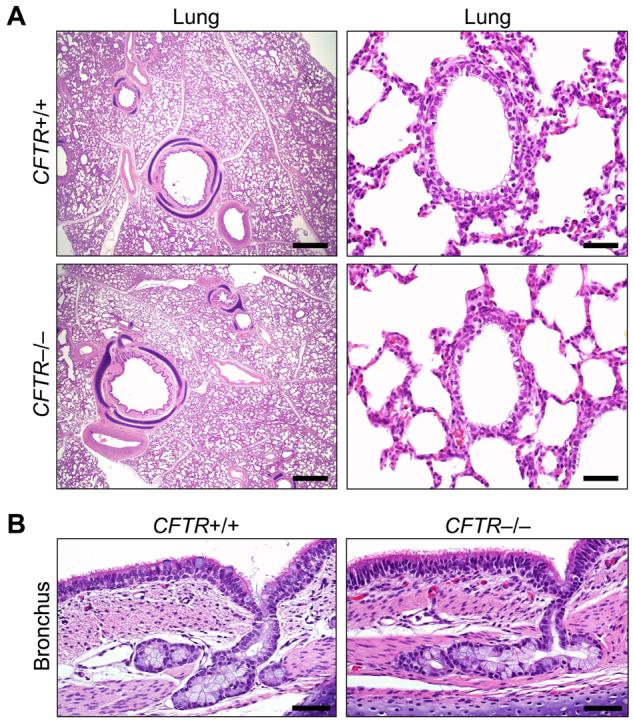
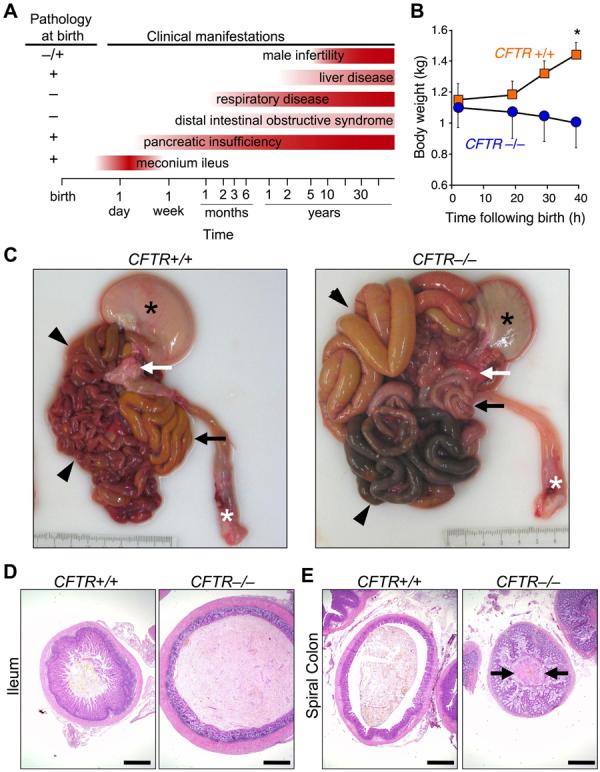
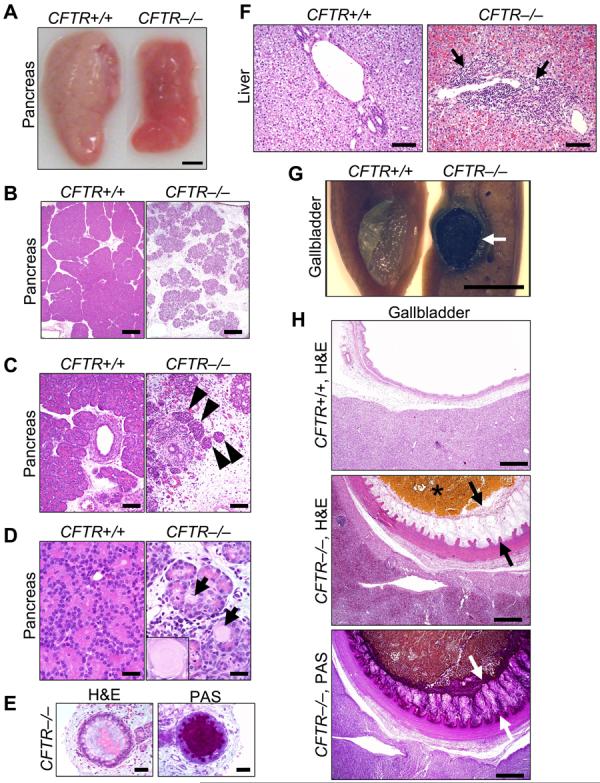
Almost two decades after CFTR was identified as the gene responsible for cystic fibrosis (CF), we still lack answers to many questions about the pathogenesis of the disease, and it remains incurable. Mice with a disrupted CFTR gene have greatly facilitated CF studies, but the mutant mice do not develop the characteristic manifestations of human CF, including abnormalities of the pancreas, lung, intestine, liver, and other organs. Because pigs share many anatomical and physiological features with humans, we generated pigs with a targeted disruption of both CFTR alleles. Newborn pigs lacking CFTR exhibited defective chloride transport and developed meconium ileus, exocrine pancreatic destruction, and focal biliary cirrhosis, replicating abnormalities seen in newborn humans with CF. The pig model may provide opportunities to address persistent questions about CF pathogenesis and accelerate discovery of strategies for prevention and treatment.
在囊性纤维化跨膜传导调节因子(CFTR)被确定为导致囊性纤维化(CF)的基因近二十年后,我们对该疾病发病机制的许多问题仍缺乏答案,并且它仍然无法治愈。CFTR基因被破坏的小鼠极大地促进了CF研究,但这些突变小鼠并未出现人类CF的典型表现,包括胰腺、肺、肠道、肝脏和其他器官的异常。由于猪与人类具有许多解剖学和生理学特征,我们培育出了CFTR两个等位基因均被靶向破坏的猪。缺乏CFTR的新生猪表现出氯化物转运缺陷,并出现胎粪性肠梗阻、外分泌胰腺破坏和局灶性胆汁性肝硬化,重现了患有CF的新生儿中所见的异常情况。猪模型可能为解决有关CF发病机制的持续问题以及加速预防和治疗策略的发现提供机会。