Böcker Sebastian, Letzel Matthias C, Lipták Zsuzsanna, Pervukhin Anton
Lehrstuhl für Bioinformatik, Friedrich-Schiller-Universität Jena, Jena, Germany.
Bioinformatics. 2009 Jan 15;25(2):218-24. doi: 10.1093/bioinformatics/btn603. Epub 2008 Nov 17.
High-resolution mass spectrometry (MS) is among the most widely used technologies in metabolomics. Metabolites participate in almost all cellular processes, but most metabolites still remain uncharacterized. Determination of the sum formula is a crucial step in the identification of an unknown metabolite, as it reduces its possible structures to a hopefully manageable set.
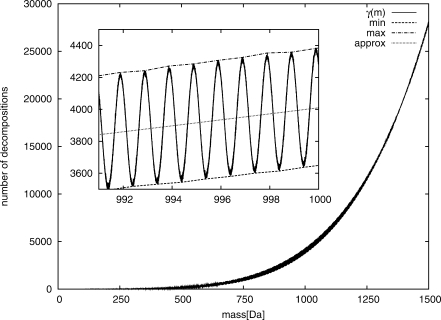
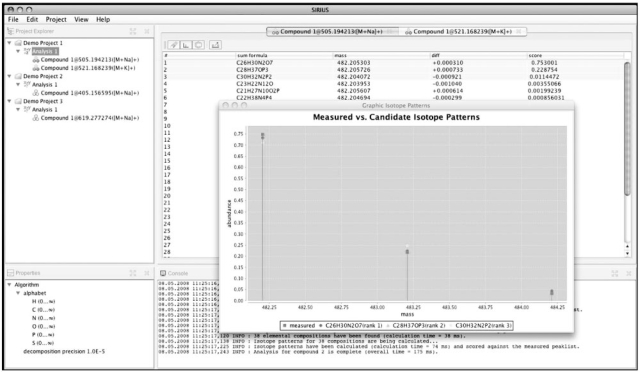
We present a method for determining the sum formula of a metabolite solely from its mass and the natural distribution of its isotopes. Our input is a measured isotope pattern from a high resolution mass spectrometer, and we want to find those molecules that best match this pattern. Our method is computationally efficient, and results on experimental data are very promising: for orthogonal time-of-flight mass spectrometry, we correctly identify sum formulas for >90% of the molecules, ranging in mass up to 1000 Da.
高分辨率质谱(MS)是代谢组学中使用最广泛的技术之一。代谢物几乎参与所有细胞过程,但大多数代谢物仍未得到表征。确定分子式是鉴定未知代谢物的关键步骤,因为它将其可能的结构减少到一个有望易于管理的集合。
我们提出了一种仅根据代谢物的质量及其同位素的自然分布来确定其分子式的方法。我们的输入是来自高分辨率质谱仪的测量同位素模式,我们希望找到最匹配此模式的那些分子。我们的方法计算效率高,实验数据的结果非常有前景:对于正交飞行时间质谱,我们正确识别了质量高达1000 Da的分子中>90%的分子式。