Lin Hong Huang, Zhang Guang Lan, Tongchusak Songsak, Reinherz Ellis L, Brusic Vladimir
Cancer Vaccine Center, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
BMC Bioinformatics. 2008 Dec 12;9 Suppl 12(Suppl 12):S22. doi: 10.1186/1471-2105-9-S12-S22.
Initiation and regulation of immune responses in humans involves recognition of peptides presented by human leukocyte antigen class II (HLA-II) molecules. These peptides (HLA-II T-cell epitopes) are increasingly important as research targets for the development of vaccines and immunotherapies. HLA-II peptide binding studies involve multiple overlapping peptides spanning individual antigens, as well as complete viral proteomes. Antigen variation in pathogens and tumor antigens, and extensive polymorphism of HLA molecules increase the number of targets for screening studies. Experimental screening methods are expensive and time consuming and reagents are not readily available for many of the HLA class II molecules. Computational prediction methods complement experimental studies, minimize the number of validation experiments, and significantly speed up the epitope mapping process. We collected test data from four independent studies that involved 721 peptide binding assays. Full overlapping studies of four antigens identified binding affinity of 103 peptides to seven common HLA-DR molecules (DRB1*0101, 0301, 0401, 0701, 1101, 1301, and 1501). We used these data to analyze performance of 21 HLA-II binding prediction servers accessible through the WWW.
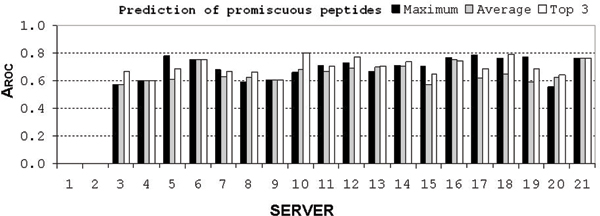
Because not all servers have predictors for all tested HLA-II molecules, we assessed a total of 113 predictors. The length of test peptides ranged from 15 to 19 amino acids. We tried three prediction strategies - the best 9-mer within the longer peptide, the average of best three 9-mer predictions, and the average of all 9-mer predictions within the longer peptide. The best strategy was the identification of a single best 9-mer within the longer peptide. Overall, measured by the receiver operating characteristic method (AROC), 17 predictors showed good (AROC > 0.8), 41 showed marginal (AROC > 0.7), and 55 showed poor performance (AROC < 0.7). Good performance predictors included HLA-DRB1*0101 (seven), 1101 (six), 0401 (three), and 0701 (one). The best individual predictor was NETMHCIIPAN, closely followed by PROPRED, IEDB (Consensus), and MULTIPRED (SVM). None of the individual predictors was shown to be suitable for prediction of promiscuous peptides. Current predictive capabilities allow prediction of only 50% of actual T-cell epitopes using practical thresholds.
The available HLA-II servers do not match prediction capabilities of HLA-I predictors. Currently available HLA-II prediction servers offer only a limited prediction accuracy and the development of improved predictors is needed for large-scale studies, such as proteome-wide epitope mapping. The requirements for accuracy of HLA-II binding predictions are stringent because of the substantial effect of false positives.

人类免疫反应的启动和调节涉及对人类白细胞抗原II类(HLA-II)分子所呈递肽段的识别。这些肽段(HLA-II T细胞表位)作为疫苗和免疫疗法开发的研究靶点,其重要性日益凸显。HLA-II肽段结合研究涉及跨越单个抗原的多个重叠肽段以及完整的病毒蛋白质组。病原体和肿瘤抗原中的抗原变异以及HLA分子的广泛多态性增加了筛选研究的靶点数量。实验筛选方法昂贵且耗时,并且许多HLA II类分子的试剂不易获得。计算预测方法可补充实验研究,减少验证实验的数量,并显著加快表位定位过程。我们从四项独立研究中收集了测试数据,这些研究涉及721次肽段结合测定。对四种抗原进行的完全重叠研究确定了103种肽段与七种常见HLA-DR分子(DRB1*0101、0301、0401、0701、1101、1301和1501)的结合亲和力。我们使用这些数据来分析通过万维网可访问的21个HLA-II结合预测服务器的性能。
由于并非所有服务器都具有针对所有测试HLA-II分子的预测器,因此我们总共评估了113个预测器。测试肽段的长度范围为15至19个氨基酸。我们尝试了三种预测策略——较长肽段内最佳的9肽、最佳三个9肽预测值的平均值以及较长肽段内所有9肽预测值的平均值。最佳策略是在较长肽段内识别单个最佳9肽。总体而言,通过接受者操作特征方法(AROC)衡量,17个预测器表现良好(AROC>0.8),41个表现中等(AROC>0.7),55个表现较差(AROC<0.7)。表现良好的预测器包括HLA-DRB1*0101(7个)、1101(6个)、0401(3个)和070(1个)。最佳的单个预测器是NETMHCIIPAN,紧随其后的是PROPRED、IEDB(共识)和MULTIPRED(支持向量机)。没有单个预测器被证明适用于预测混杂肽段。当前的预测能力使用实际阈值仅能预测50%的实际T细胞表位。
现有的HLA-II服务器与HLA-I预测器的预测能力不匹配。目前可用的HLA-II预测服务器仅提供有限的预测准确性,对于大规模研究,如全蛋白质组表位定位,需要开发改进的预测器。由于假阳性的实质性影响,对HLA-II结合预测准确性的要求很严格。