Bradley Robert K, Roberts Adam, Smoot Michael, Juvekar Sudeep, Do Jaeyoung, Dewey Colin, Holmes Ian, Pachter Lior
Department of Mathematics, University of California Berkeley, Berkeley, California, United States of America.
PLoS Comput Biol. 2009 May;5(5):e1000392. doi: 10.1371/journal.pcbi.1000392. Epub 2009 May 29.
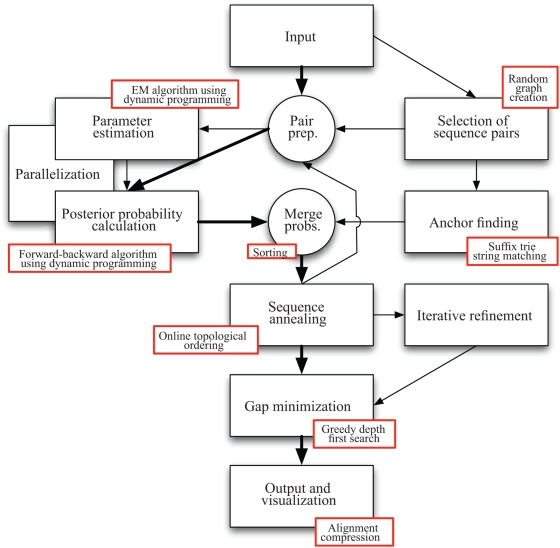
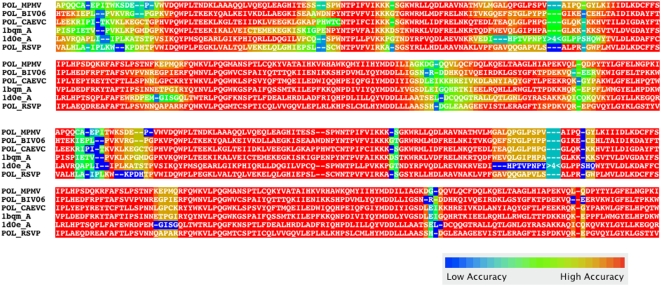
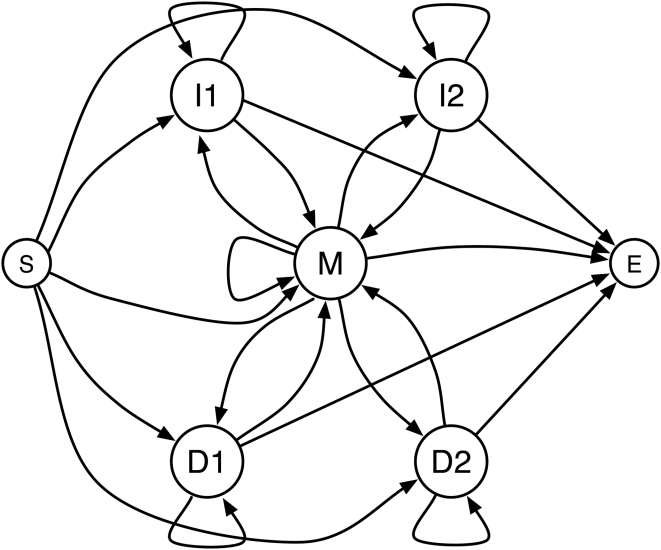
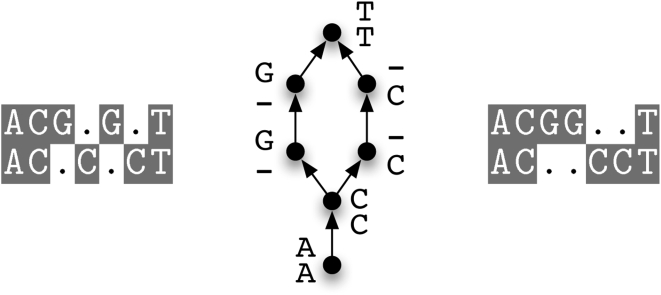
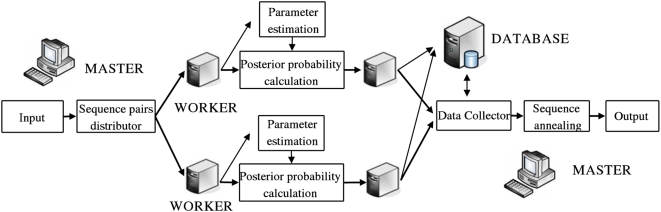
We describe a new program for the alignment of multiple biological sequences that is both statistically motivated and fast enough for problem sizes that arise in practice. Our Fast Statistical Alignment program is based on pair hidden Markov models which approximate an insertion/deletion process on a tree and uses a sequence annealing algorithm to combine the posterior probabilities estimated from these models into a multiple alignment. FSA uses its explicit statistical model to produce multiple alignments which are accompanied by estimates of the alignment accuracy and uncertainty for every column and character of the alignment--previously available only with alignment programs which use computationally-expensive Markov Chain Monte Carlo approaches--yet can align thousands of long sequences. Moreover, FSA utilizes an unsupervised query-specific learning procedure for parameter estimation which leads to improved accuracy on benchmark reference alignments in comparison to existing programs. The centroid alignment approach taken by FSA, in combination with its learning procedure, drastically reduces the amount of false-positive alignment on biological data in comparison to that given by other methods. The FSA program and a companion visualization tool for exploring uncertainty in alignments can be used via a web interface at http://orangutan.math.berkeley.edu/fsa/, and the source code is available at http://fsa.sourceforge.net/.
我们描述了一种用于多生物序列比对的新程序,该程序既具有统计学依据,又对于实际中出现的问题规模而言足够快速。我们的快速统计比对程序基于成对隐马尔可夫模型,该模型近似于树上的插入/删除过程,并使用序列退火算法将从这些模型估计的后验概率组合成一个多序列比对。FSA 使用其明确的统计模型来生成多序列比对,同时为比对的每一列和每个字符提供比对准确性和不确定性的估计——此前只有使用计算成本高昂的马尔可夫链蒙特卡罗方法的比对程序才能做到这一点——而且能够比对数千条长序列。此外,FSA 利用一种无监督的特定查询学习程序进行参数估计,与现有程序相比,这使得在基准参考比对上的准确性得到提高。FSA 采用的质心比对方法及其学习程序,与其他方法相比,极大地减少了生物数据上的假阳性比对数量。FSA 程序以及一个用于探索比对不确定性的配套可视化工具可通过网页界面 http://orangutan.math.berkeley.edu/fsa/ 使用,其源代码可在 http://fsa.sourceforge.net/ 获取。