Maruyama Fumito, Kobata Mitsuhiko, Kurokawa Ken, Nishida Keishin, Sakurai Atsuo, Nakano Kazuhiko, Nomura Ryota, Kawabata Shigetada, Ooshima Takashi, Nakai Kenta, Hattori Masahira, Hamada Shigeyuki, Nakagawa Ichiro
Division of Bacteriology, Department of Infectious Diseases Control, International Research Center for Infectious Diseases, The Institute of Medical Science, The University of Tokyo, Tokyo 108-8639, Japan.
BMC Genomics. 2009 Aug 5;10:358. doi: 10.1186/1471-2164-10-358.
Streptococcus mutans is the major pathogen of dental caries, and it occasionally causes infective endocarditis. While the pathogenicity of this species is distinct from other human pathogenic streptococci, the species-specific evolution of the genus Streptococcus and its genomic diversity are poorly understood.
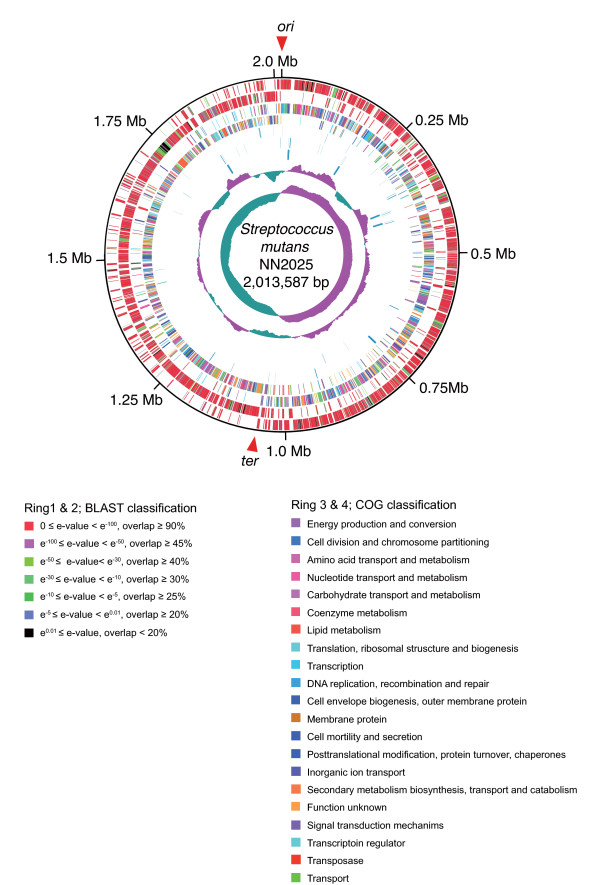
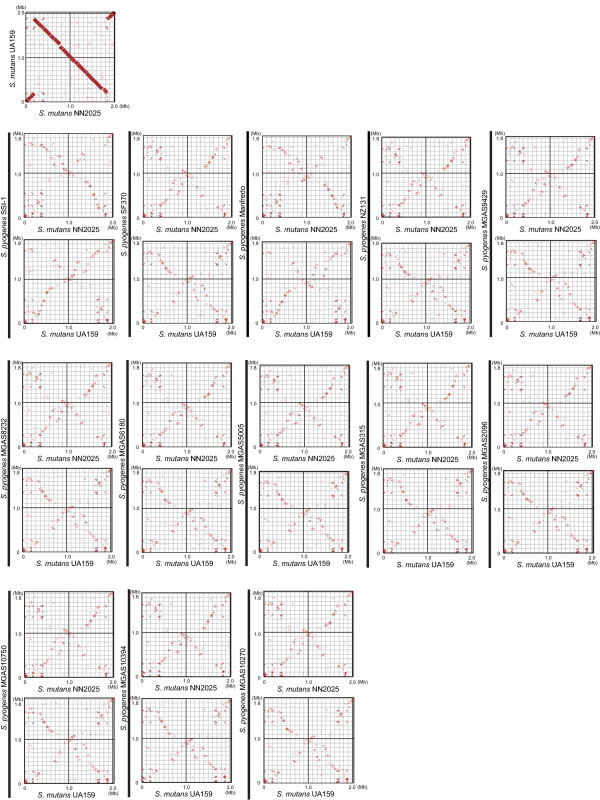
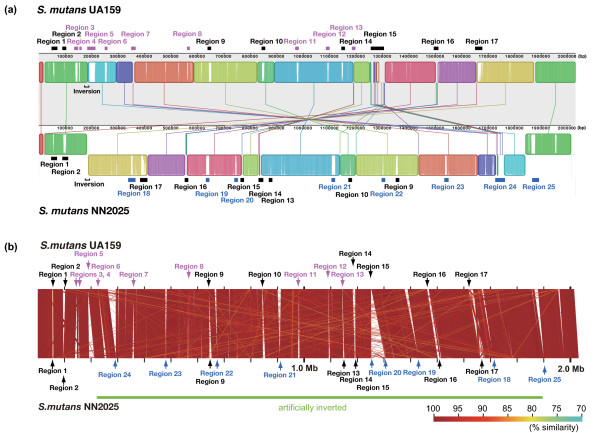
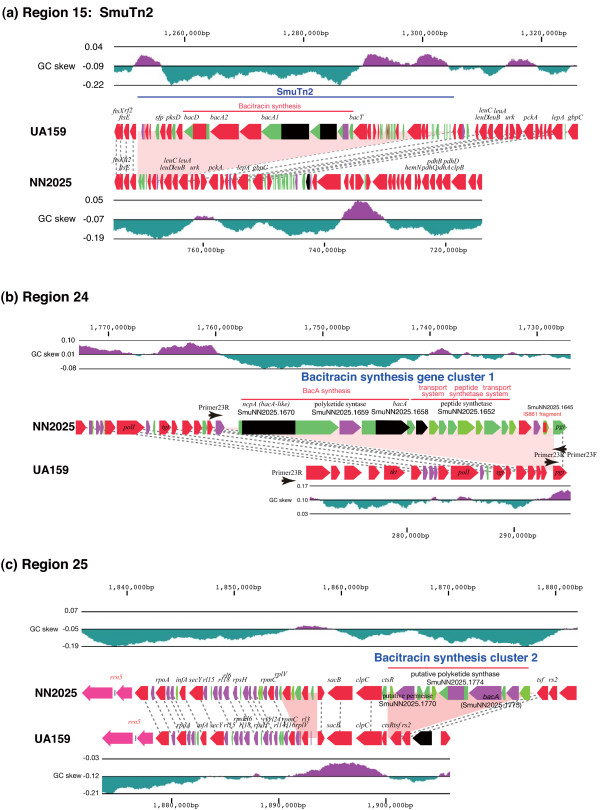
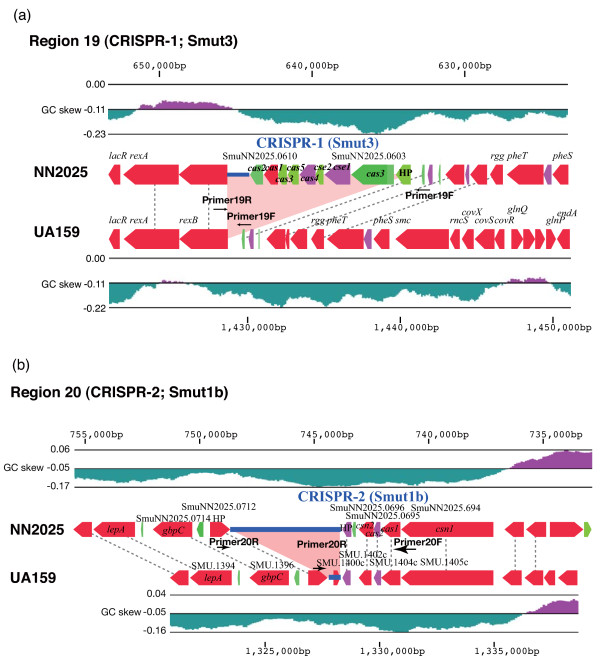
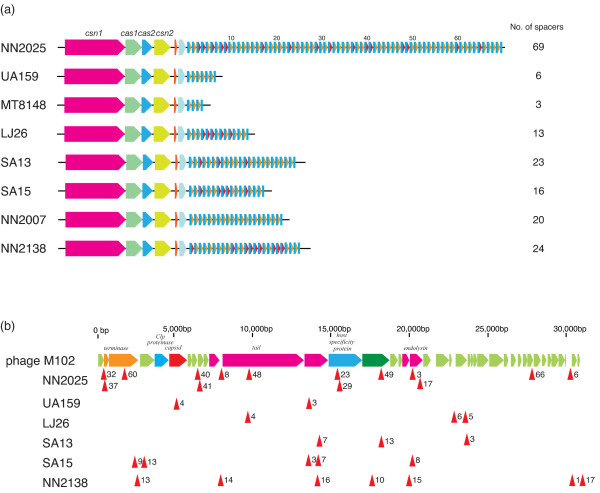
We have sequenced the complete genome of S. mutans serotype c strain NN2025, and compared it with the genome of UA159. The NN2025 genome is composed of 2,013,587 bp, and the two strains show highly conserved core-genome. However, comparison of the two S. mutans strains showed a large genomic inversion across the replication axis producing an X-shaped symmetrical DNA dot plot. This phenomenon was also observed between other streptococcal species, indicating that streptococcal genetic rearrangements across the replication axis play an important role in Streptococcus genetic shuffling. We further confirmed the genomic diversity among 95 clinical isolates using long-PCR analysis. Genomic diversity in S. mutans appears to occur frequently between insertion sequence (IS) elements and transposons, and these diversity regions consist of restriction/modification systems, antimicrobial peptide synthesis systems, and transporters. S. mutans may preferentially reject the phage infection by clustered regularly interspaced short palindromic repeats (CRISPRs). In particular, the CRISPR-2 region, which is highly divergent between strains, in NN2025 has long repeated spacer sequences corresponding to the streptococcal phage genome.
These observations suggest that S. mutans strains evolve through chromosomal shuffling and that phage infection is not needed for gene acquisition. In contrast, S. pyogenes tolerates phage infection for acquisition of virulence determinants for niche adaptation.
变形链球菌是龋齿的主要病原体,偶尔也会引起感染性心内膜炎。虽然该菌种的致病性与其他人类致病链球菌不同,但对链球菌属的种特异性进化及其基因组多样性了解甚少。
我们对变形链球菌血清型c菌株NN2025的全基因组进行了测序,并与UA159的基因组进行了比较。NN2025基因组由2,013,587 bp组成,这两个菌株显示出高度保守的核心基因组。然而,对这两种变形链球菌菌株的比较显示,在复制轴上存在一个大的基因组倒位,产生了一个X形对称的DNA点阵图。在其他链球菌物种之间也观察到了这种现象,表明跨复制轴的链球菌基因重排在链球菌基因改组中起重要作用。我们使用长PCR分析进一步证实了95株临床分离株之间的基因组多样性。变形链球菌的基因组多样性似乎经常发生在插入序列(IS)元件和转座子之间,这些多样性区域由限制/修饰系统、抗菌肽合成系统和转运蛋白组成。变形链球菌可能通过成簇规律间隔短回文重复序列(CRISPRs)优先排斥噬菌体感染。特别是,NN2025中菌株间高度不同的CRISPR-2区域具有与链球菌噬菌体基因组相对应的长重复间隔序列。
这些观察结果表明,变形链球菌菌株通过染色体改组进化,并且基因获取不需要噬菌体感染。相比之下,化脓性链球菌耐受噬菌体感染以获取适应生态位的毒力决定因素。