Department of Statistics, University of Wisconsin - Madison, Madison, WI 53706, USA.
Bioinformatics. 2009 Nov 1;25(21):2780-6. doi: 10.1093/bioinformatics/btp502. Epub 2009 Aug 18.
The power of a microarray experiment derives from the identification of genes differentially regulated across biological conditions. To date, differential regulation is most often taken to mean differential expression, and a number of useful methods for identifying differentially expressed (DE) genes or gene sets are available. However, such methods are not able to identify many relevant classes of differentially regulated genes. One important example concerns differentially co-expressed (DC) genes.
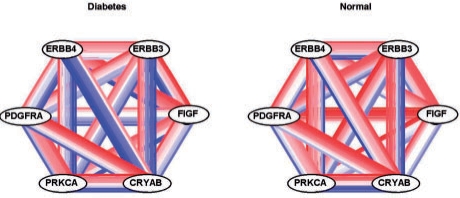
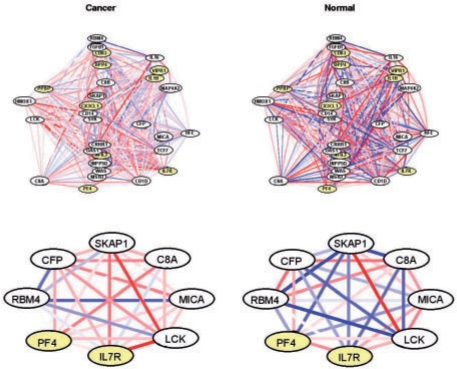
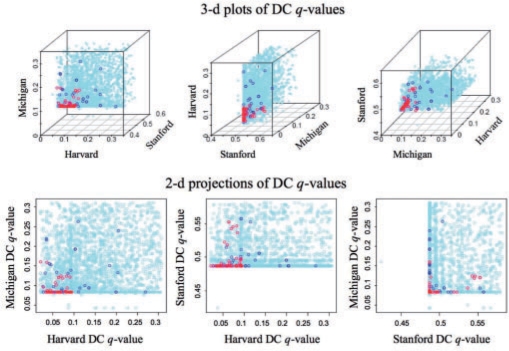
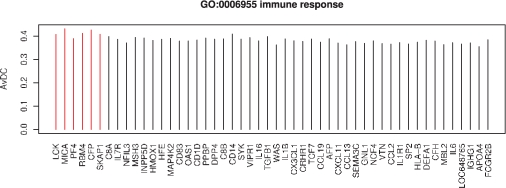
We propose an approach, gene set co-expression analysis (GSCA), to identify DC gene sets. The GSCA approach provides a false discovery rate controlled list of interesting gene sets, does not require that genes be highly correlated in at least one biological condition and is readily applied to data from individual or multiple experiments, as we demonstrate using data from studies of lung cancer and diabetes.
The GSCA approach is implemented in R and available at www.biostat.wisc.edu/ approximately kendzior/GSCA/.
Supplementary data are available at Bioinformatics online.
微阵列实验的威力源自于识别在不同生物学条件下差异调控的基因。迄今为止,差异调控通常意味着差异表达,并且有许多有用的方法可用于识别差异表达(DE)基因或基因集。但是,这些方法无法识别许多相关的差异调控基因类别。一个重要的例子涉及差异共表达(DC)基因。
我们提出了一种方法,即基因集共表达分析(GSCA),用于识别 DC 基因集。GSCA 方法提供了一个具有控制假发现率的有趣基因集列表,不需要基因在至少一种生物学条件下高度相关,并且易于应用于单个或多个实验的数据,我们使用来自肺癌和糖尿病研究的数据证明了这一点。
GSCA 方法在 R 中实现,并可在 www.biostat.wisc.edu/approximately/kendzior/GSCA/ 上获得。
补充数据可在 Bioinformatics 在线获得。