Leng Ning, Li Yuan, McIntosh Brian E, Nguyen Bao Kim, Duffin Bret, Tian Shulan, Thomson James A, Dewey Colin N, Stewart Ron, Kendziorski Christina
Department of Statistics, University of Wisconsin, Madison, WI, USA, Regenerative Biology, Morgridge Institute for Research, Madison, WI, USA.
Department of Statistics, University of Wisconsin, Madison, WI, USA.
Bioinformatics. 2015 Aug 15;31(16):2614-22. doi: 10.1093/bioinformatics/btv193. Epub 2015 Apr 5.
With improvements in next-generation sequencing technologies and reductions in price, ordered RNA-seq experiments are becoming common. Of primary interest in these experiments is identifying genes that are changing over time or space, for example, and then characterizing the specific expression changes. A number of robust statistical methods are available to identify genes showing differential expression among multiple conditions, but most assume conditions are exchangeable and thereby sacrifice power and precision when applied to ordered data.
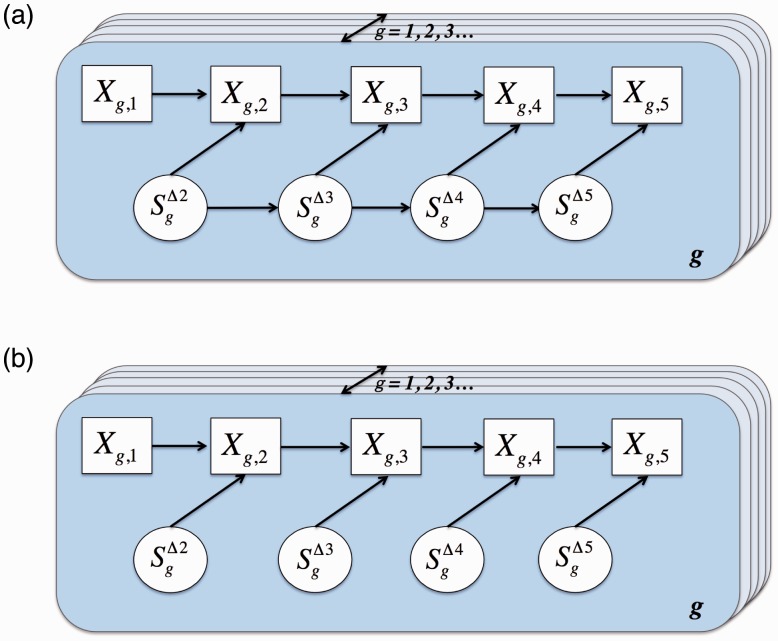
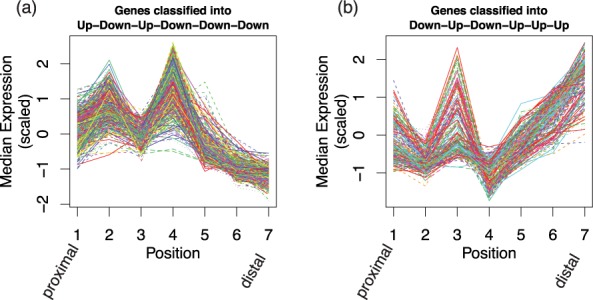
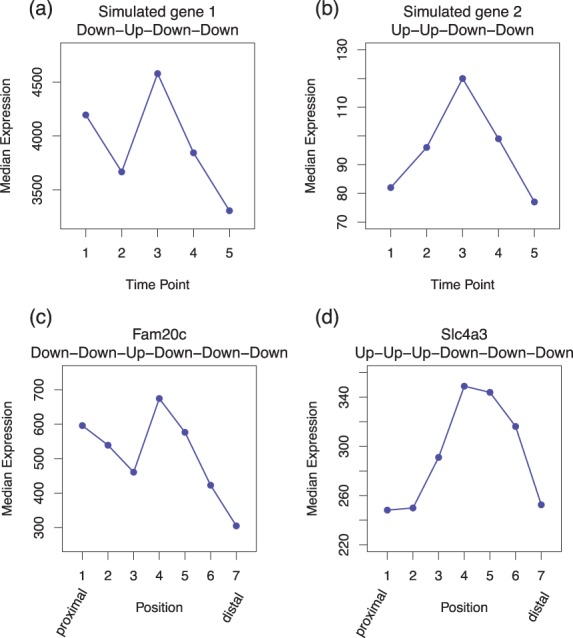
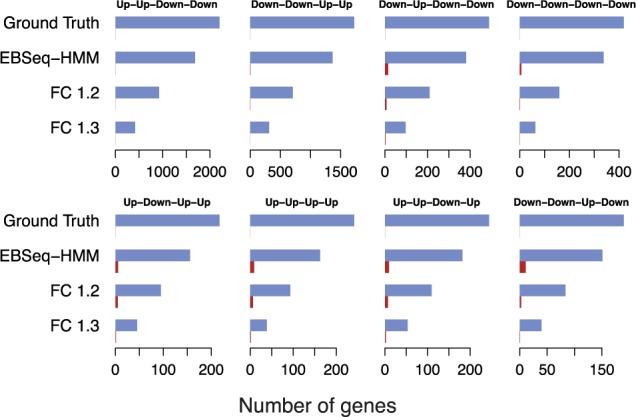
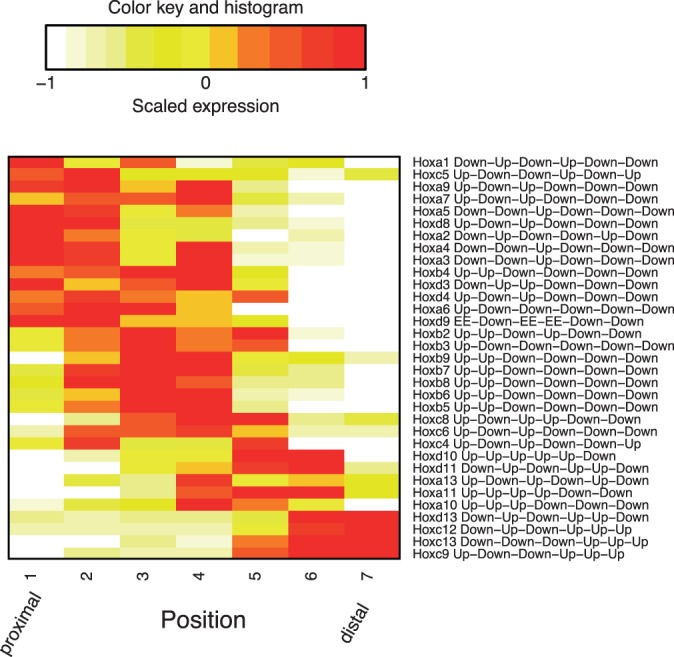
We propose an empirical Bayes mixture modeling approach called EBSeq-HMM. In EBSeq-HMM, an auto-regressive hidden Markov model is implemented to accommodate dependence in gene expression across ordered conditions. As demonstrated in simulation and case studies, the output proves useful in identifying differentially expressed genes and in specifying gene-specific expression paths. EBSeq-HMM may also be used for inference regarding isoform expression.
An R package containing examples and sample datasets is available at Bioconductor.
Supplementary data are available at Bioinformatics online.
随着下一代测序技术的改进和价格的降低,有序RNA测序实验变得越来越普遍。例如,这些实验的主要兴趣在于识别随时间或空间变化的基因,然后表征特定的表达变化。有许多强大的统计方法可用于识别在多种条件下显示差异表达的基因,但大多数方法假设条件是可交换的,因此在应用于有序数据时会牺牲功效和精度。
我们提出了一种称为EBSeq-HMM的经验贝叶斯混合建模方法。在EBSeq-HMM中,实现了一个自回归隐马尔可夫模型,以适应跨有序条件的基因表达依赖性。如模拟和案例研究所示,结果证明在识别差异表达基因和指定基因特异性表达路径方面很有用。EBSeq-HMM也可用于推断异构体表达。
在Bioconductor上提供了一个包含示例和样本数据集的R包。
补充数据可在《生物信息学》在线获取。