Hafner Marc, Koeppl Heinz, Hasler Martin, Wagner Andreas
School of Computer and Communication Sciences, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
PLoS Comput Biol. 2009 Oct;5(10):e1000534. doi: 10.1371/journal.pcbi.1000534. Epub 2009 Oct 16.
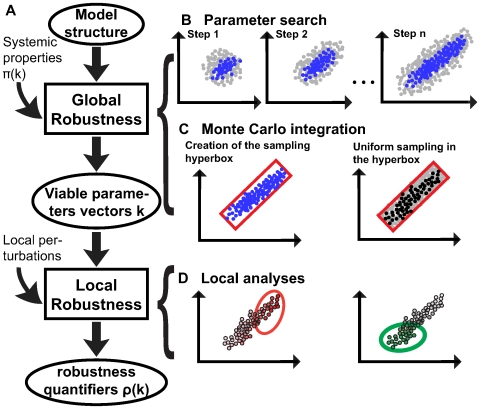
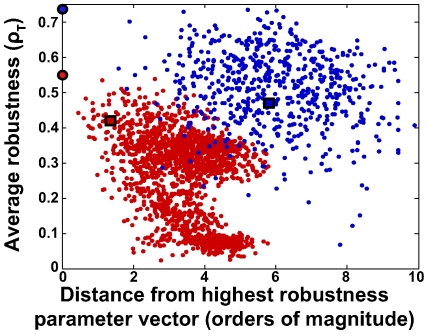
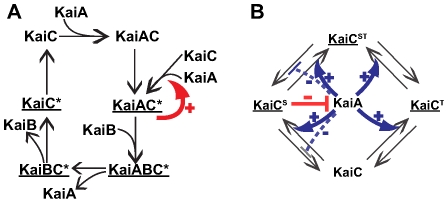
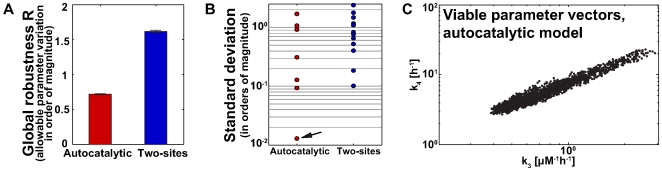
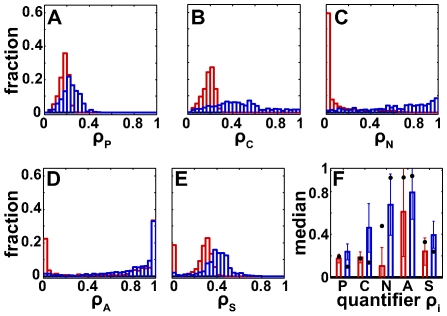
To characterize the behavior and robustness of cellular circuits with many unknown parameters is a major challenge for systems biology. Its difficulty rises exponentially with the number of circuit components. We here propose a novel analysis method to meet this challenge. Our method identifies the region of a high-dimensional parameter space where a circuit displays an experimentally observed behavior. It does so via a Monte Carlo approach guided by principal component analysis, in order to allow efficient sampling of this space. This 'global' analysis is then supplemented by a 'local' analysis, in which circuit robustness is determined for each of the thousands of parameter sets sampled in the global analysis. We apply this method to two prominent, recent models of the cyanobacterial circadian oscillator, an autocatalytic model, and a model centered on consecutive phosphorylation at two sites of the KaiC protein, a key circadian regulator. For these models, we find that the two-sites architecture is much more robust than the autocatalytic one, both globally and locally, based on five different quantifiers of robustness, including robustness to parameter perturbations and to molecular noise. Our 'glocal' combination of global and local analyses can also identify key causes of high or low robustness. In doing so, our approach helps to unravel the architectural origin of robust circuit behavior. Complementarily, identifying fragile aspects of system behavior can aid in designing perturbation experiments that may discriminate between competing mechanisms and different parameter sets.
表征具有许多未知参数的细胞回路的行为和稳健性是系统生物学面临的一项重大挑战。其难度会随着回路组件数量呈指数级增加。我们在此提出一种新颖的分析方法来应对这一挑战。我们的方法可识别高维参数空间中回路呈现实验观测行为的区域。它通过主成分分析引导的蒙特卡罗方法来实现这一点,以便对该空间进行高效采样。这种“全局”分析随后会辅以“局部”分析,其中会针对在全局分析中采样的数千个参数集分别确定回路的稳健性。我们将此方法应用于两个著名的近期蓝藻生物钟振荡器模型,一个自催化模型,以及一个以关键生物钟调节蛋白KaiC的两个位点连续磷酸化为核心的模型。对于这些模型,基于包括对参数扰动和分子噪声的稳健性在内的五种不同稳健性量化指标,我们发现双位点结构在全局和局部上都比自催化结构稳健得多。我们的全局和局部分析的“全局 - 局部”组合还可以识别高稳健性或低稳健性的关键原因。通过这样做,我们的方法有助于揭示稳健回路行为的架构起源。作为补充,识别系统行为的脆弱方面有助于设计扰动实验,这些实验可能区分相互竞争的机制和不同的参数集。