Pušnik Žiga, Mraz Miha, Zimic Nikolaj, Moškon Miha
University of Ljubljana, Faculty of Computer and Information Science, Večna pot 113, Ljubljana, 1000 Slovenia.
J Biol Eng. 2019 Sep 18;13:75. doi: 10.1186/s13036-019-0205-0. eCollection 2019.
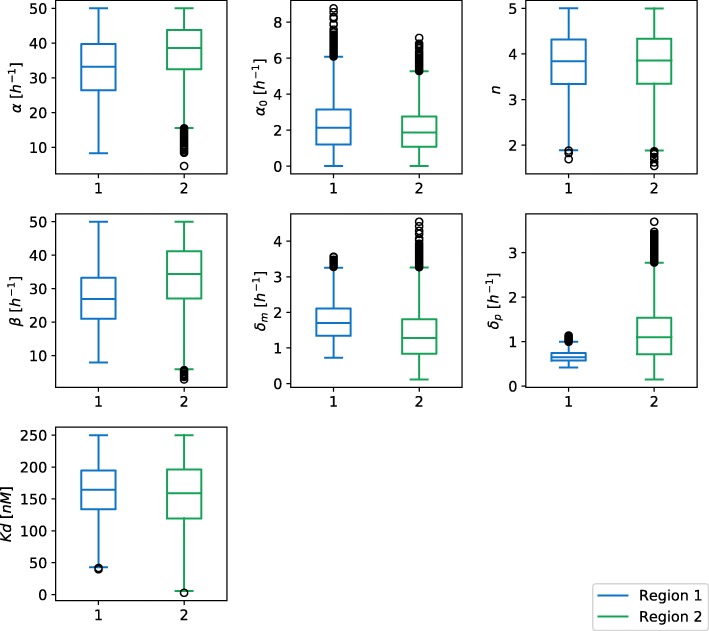
Gene regulatory networks with different topological and/or dynamical properties might exhibit similar behavior. System that is less perceptive for the perturbations of its internal and external factors should be preferred. Methods for sensitivity and robustness assessment have already been developed and can be roughly divided into local and global approaches. Local methods focus only on the local area around nominal parameter values. This can be problematic when parameters exhibits the desired behavior over a large range of parameter perturbations or when parameter values are unknown. Global methods, on the other hand, investigate the whole space of parameter values and mostly rely on different sampling techniques. This can be computationally inefficient. To address these shortcomings 'glocal' approaches were developed that apply global and local approaches in an effective and rigorous manner.
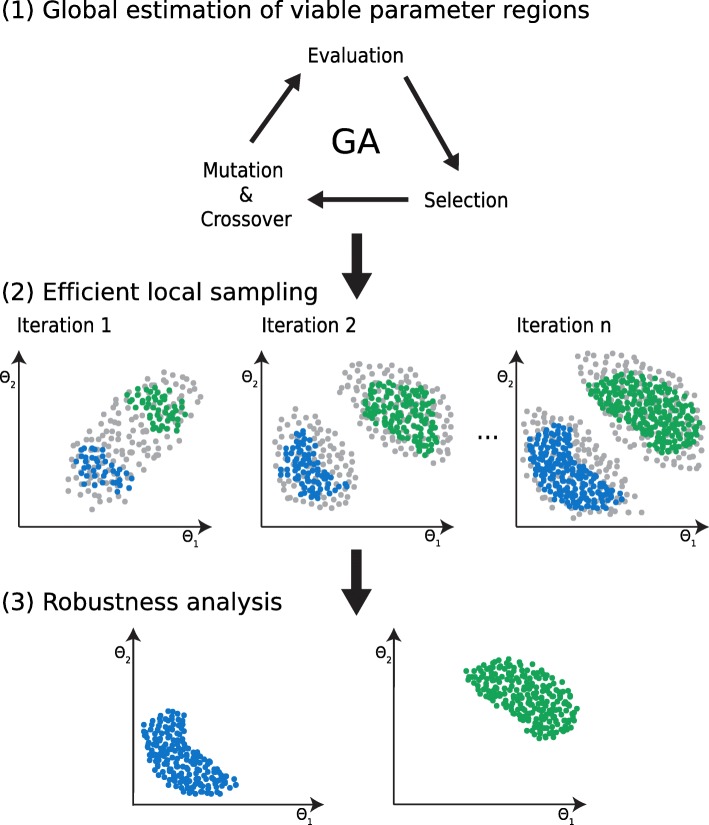
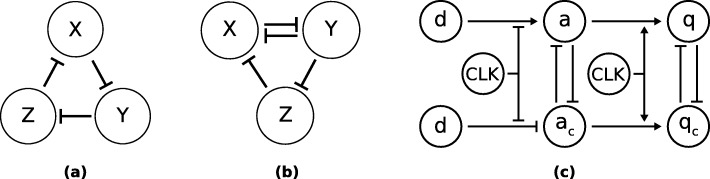
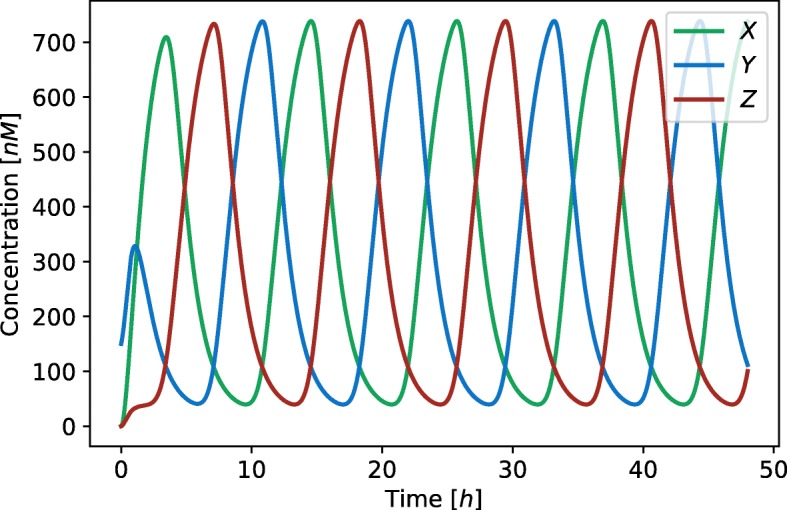
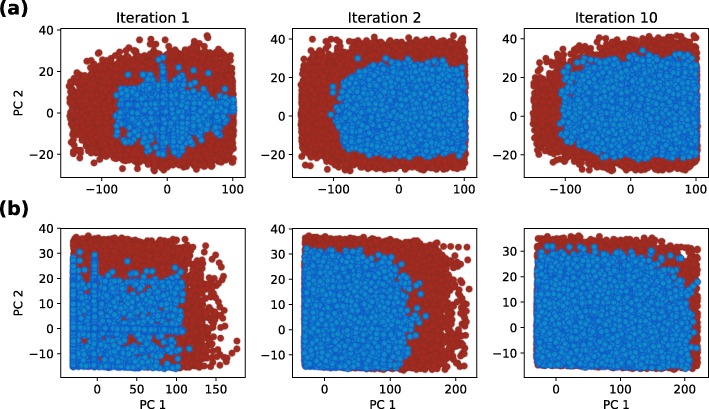
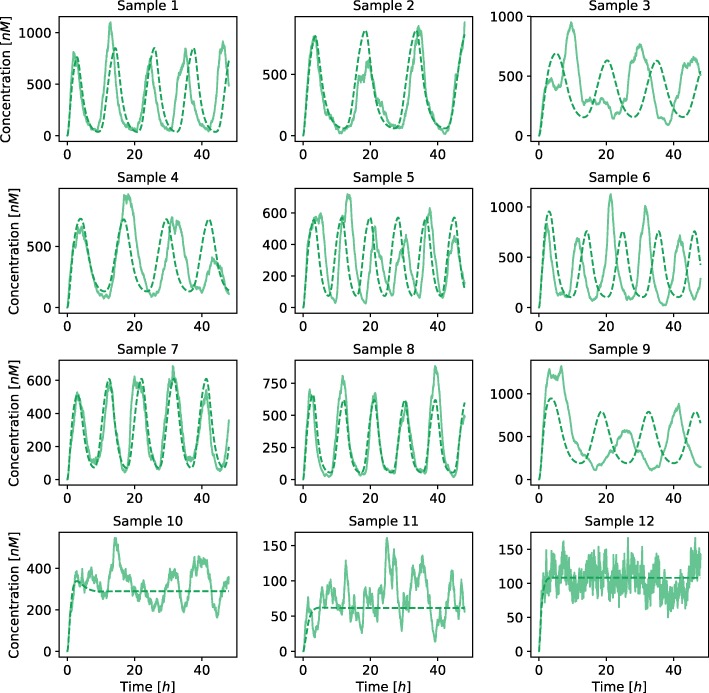
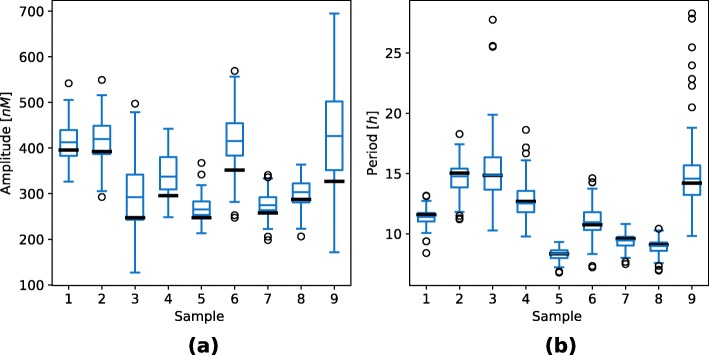
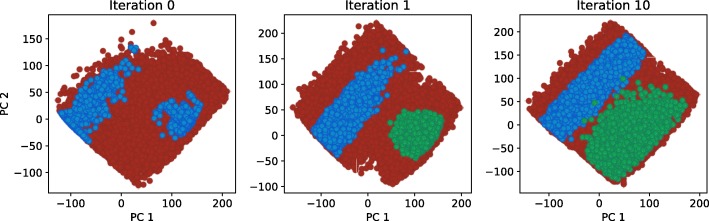
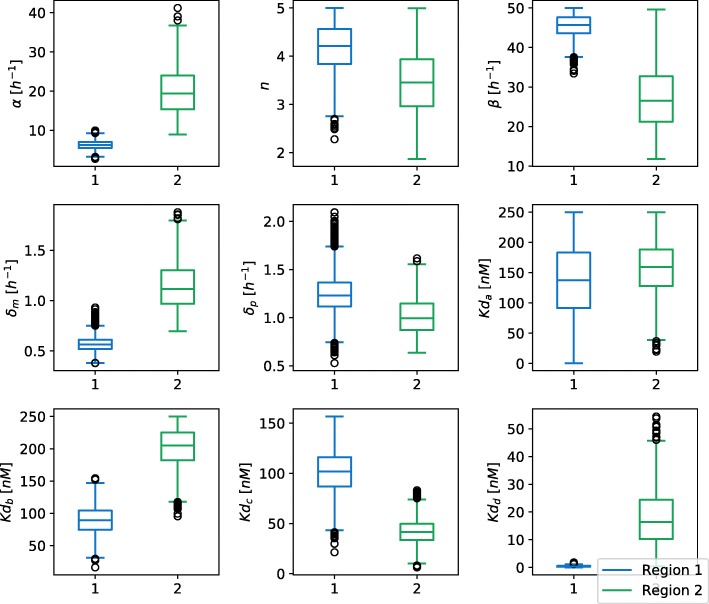
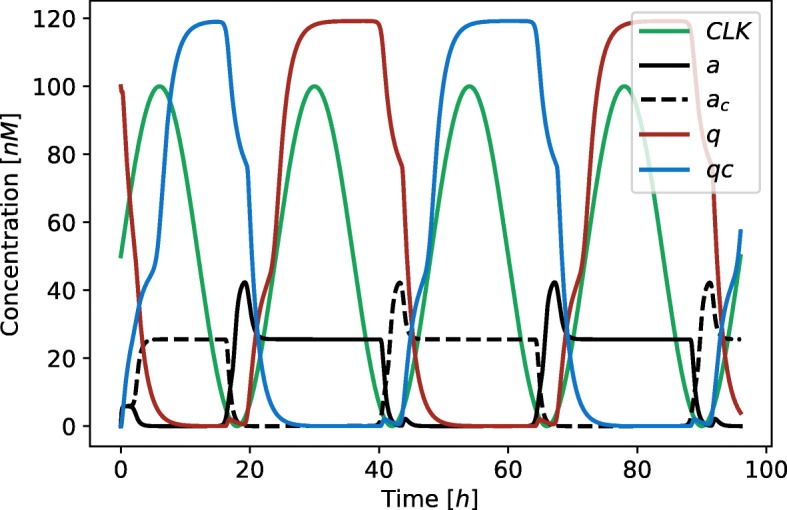
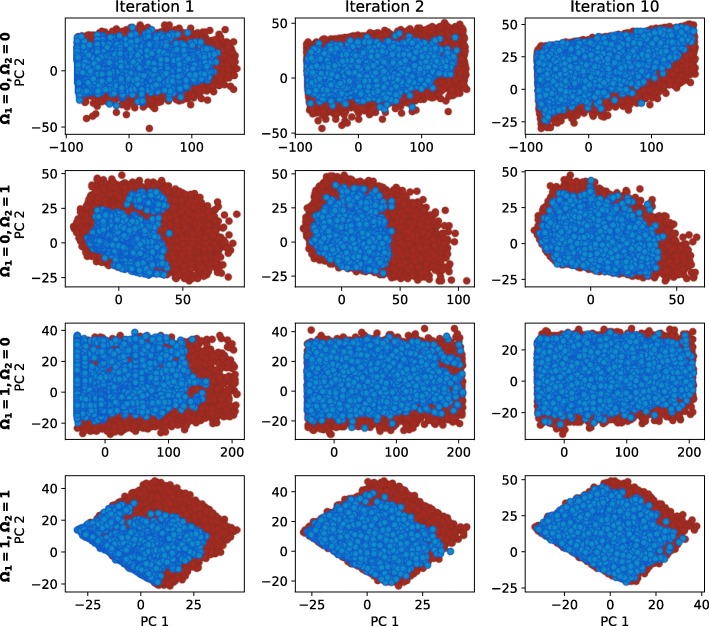
Herein, we present a computational approach for 'glocal' analysis of viable parameter regions in biological models. The methodology is based on the exploration of high-dimensional viable parameter spaces with global and local sampling, clustering and dimensionality reduction techniques. The proposed methodology allows us to efficiently investigate the viable parameter space regions, evaluate the regions which exhibit the largest robustness, and to gather new insights regarding the size and connectivity of the viable parameter regions. We evaluate the proposed methodology on three different synthetic gene regulatory network models, i.e. the repressilator model, the model of the AC-DC circuit and the model of the edge-triggered master-slave D flip-flop.
The proposed methodology provides a rigorous assessment of the shape and size of viable parameter regions based on (1) the mathematical description of the biological system of interest, (2) constraints that define feasible parameter regions and (3) cost function that defines the desired or observed behavior of the system. These insights can be used to assess the robustness of biological systems, even in the case when parameter values are unknown and more importantly, even when there are multiple poorly connected viable parameter regions in the solution space. Moreover, the methodology can be efficiently applied to the analysis of biological systems that exhibit multiple modes of the targeted behavior.
具有不同拓扑和/或动力学特性的基因调控网络可能表现出相似的行为。应优先选择对其内部和外部因素扰动不太敏感的系统。已经开发出了灵敏度和稳健性评估方法,大致可分为局部方法和全局方法。局部方法仅关注标称参数值周围的局部区域。当参数在大范围的参数扰动下表现出期望的行为时,或者当参数值未知时,这可能会出现问题。另一方面,全局方法研究参数值的整个空间,并且大多依赖于不同的采样技术。这在计算上可能效率低下。为了解决这些缺点,开发了“全局 - 局部”方法,该方法以有效且严格的方式应用全局和局部方法。
在此,我们提出一种用于对生物模型中可行参数区域进行“全局 - 局部”分析的计算方法。该方法基于利用全局和局部采样、聚类及降维技术对高维可行参数空间进行探索。所提出的方法使我们能够有效地研究可行参数空间区域,评估表现出最大稳健性的区域,并获得有关可行参数区域的大小和连通性的新见解。我们在三个不同的合成基因调控网络模型上评估了所提出的方法,即阻遏物模型、交流 - 直流电路模型和边沿触发主从D触发器模型。
所提出的方法基于(1)感兴趣的生物系统的数学描述、(2)定义可行参数区域的约束条件以及(3)定义系统期望或观察到的行为的成本函数,对可行参数区域的形状和大小进行了严格评估。即使在参数值未知的情况下,更重要的是,即使在解空间中存在多个连接不佳的可行参数区域时,这些见解也可用于评估生物系统的稳健性。此外,该方法可以有效地应用于对表现出多种目标行为模式的生物系统的分析。