USDA/ARS National Center for Cool and Cold Water Aquaculture, Leetown, West Virginia 25430, USA.
BMC Genet. 2009 Dec 14;10:83. doi: 10.1186/1471-2156-10-83.
The use of molecular genetic technologies for broodstock management and selective breeding of aquaculture species is becoming increasingly more common with the continued development of genome tools and reagents. Several laboratories have produced genetic maps for rainbow trout to aid in the identification of loci affecting phenotypes of interest. These maps have resulted in the identification of many quantitative/qualitative trait loci affecting phenotypic variation in traits associated with albinism, disease resistance, temperature tolerance, sex determination, embryonic development rate, spawning date, condition factor and growth. Unfortunately, the elucidation of the precise allelic variation and/or genes underlying phenotypic diversity has yet to be achieved in this species having low marker densities and lacking a whole genome reference sequence. Experimental designs which integrate segregation analyses with linkage disequilibrium (LD) approaches facilitate the discovery of genes affecting important traits. To date the extent of LD has been characterized for humans and several agriculturally important livestock species but not for rainbow trout.
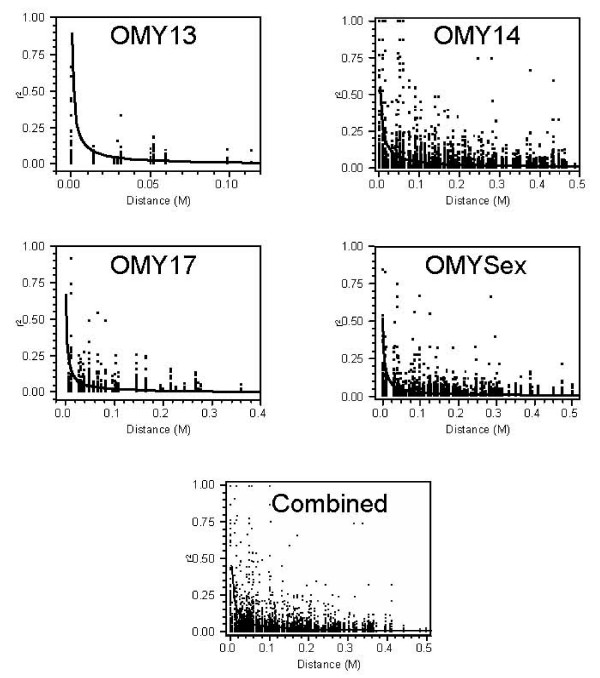
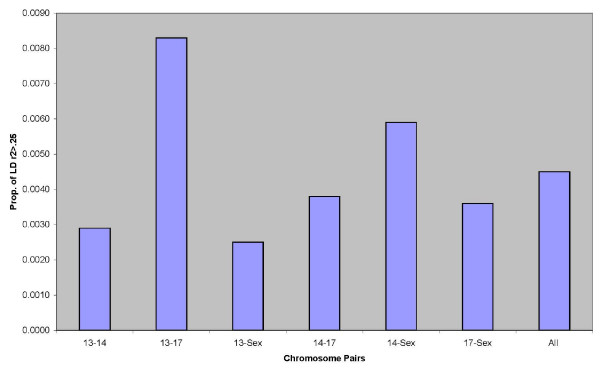
We observed that the level of LD between syntenic loci decayed rapidly at distances greater than 2 cM which is similar to observations of LD in other agriculturally important species including cattle, sheep, pigs and chickens. However, in some cases significant LD was also observed up to 50 cM. Our estimate of effective population size based on genome wide estimates of LD for the NCCCWA broodstock population was 145, indicating that this population will respond well to high selection intensity. However, the range of effective population size based on individual chromosomes was 75.51 - 203.35, possibly indicating that suites of genes on each chromosome are disproportionately under selection pressures.
Our results indicate that large numbers of markers, more than are currently available for this species, will be required to enable the use of genome-wide integrated mapping approaches aimed at identifying genes of interest in rainbow trout.
随着基因组工具和试剂的不断发展,分子遗传技术在水产养殖物种的亲鱼管理和选择性育种中的应用越来越普遍。几家实验室已经为虹鳟鱼制作了遗传图谱,以帮助确定影响感兴趣表型的基因座。这些图谱导致了许多数量/质量性状基因座的鉴定,这些基因座影响与白化病、抗病性、温度耐受性、性别决定、胚胎发育速度、产卵日期、条件系数和生长相关的性状的表型变异。不幸的是,在这个标记密度低且缺乏全基因组参考序列的物种中,尚未阐明表型多样性的精确等位基因变异和/或基因。将分离分析与连锁不平衡(LD)方法相结合的实验设计有助于发现影响重要性状的基因。迄今为止,LD 的程度已经在人类和几种重要的农业家畜物种中进行了描述,但尚未在虹鳟鱼中进行描述。
我们观察到,在距离大于 2cM 的情况下,同线基因座之间的 LD 水平迅速衰减,这与其他重要农业物种(包括牛、羊、猪和鸡)的 LD 观察结果相似。然而,在某些情况下,在多达 50cM 的距离上也观察到了显著的 LD。我们基于 NCCCWA 亲鱼群体全基因组 LD 的估计得出的有效种群大小为 145,表明该群体将很好地响应高强度的选择。然而,基于单个染色体的有效种群大小范围为 75.51-203.35,这可能表明每个染色体上的基因座套件受到选择压力的不成比例。
我们的结果表明,需要大量的标记,比目前该物种可用的标记多,才能利用旨在鉴定虹鳟鱼中感兴趣基因的全基因组综合图谱方法。