Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, Oxford, UK.
Genet Epidemiol. 2010 May;34(4):319-26. doi: 10.1002/gepi.20482.
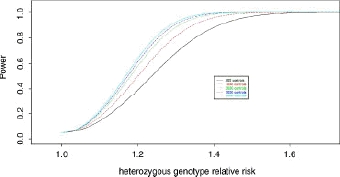
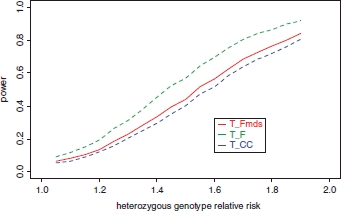
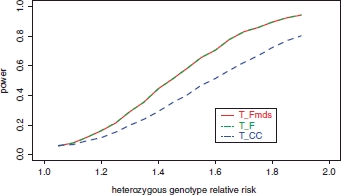
Genome-wide association (GWA) studies have proved extremely successful in identifying novel genetic loci contributing effects to complex human diseases. In doing so, they have highlighted the fact that many potential loci of modest effect remain undetected, partly due to the need for samples consisting of many thousands of individuals. Large-scale international initiatives, such as the Wellcome Trust Case Control Consortium, the Genetic Association Information Network, and the database of genetic and phenotypic information, aim to facilitate discovery of modest-effect genes by making genome-wide data publicly available, allowing information to be combined for the purpose of pooled analysis. In principle, disease or control samples from these studies could be used to increase the power of any GWA study via judicious use as "genetically matched controls" for other traits. Here, we present the biological motivation for the problem and the theoretical potential for expanding the control group with publicly available disease or reference samples. We demonstrate that a naïve application of this strategy can greatly inflate the false-positive error rate in the presence of population structure. As a remedy, we make use of genome-wide data and model selection techniques to identify "axes" of genetic variation which are associated with disease. These axes are then included as covariates in association analysis to correct for population structure, which can result in increases in power over standard analysis of genetic information from the samples in the original GWA study.
全基因组关联 (GWA) 研究已被证明在确定对复杂人类疾病有影响的新遗传基因座方面非常成功。通过这样做,它们突出了一个事实,即许多潜在的、效应适中的基因座仍然未被发现,部分原因是需要由数千个人组成的样本。大型国际倡议,如威康信托基金会病例对照联合会、遗传关联信息网络和基因与表型信息数据库,旨在通过公开提供全基因组数据,允许为合并分析而合并信息,来促进发现效应适中的基因。原则上,可以使用这些研究中的疾病或对照样本,通过明智地将其用作其他特征的“基因匹配对照”,来增加任何 GWA 研究的功效。在这里,我们提出了该问题的生物学动机和利用公开的疾病或参考样本扩展对照组的理论潜力。我们证明,在存在群体结构的情况下,这种策略的盲目应用会极大地增加假阳性错误率。作为补救措施,我们利用全基因组数据和模型选择技术来识别与疾病相关的遗传变异“轴”。然后,将这些轴作为协变量包含在关联分析中,以纠正群体结构,这可以提高对原始 GWA 研究中样本的遗传信息进行标准分析的功效。