Departamento de Ingeniería Celular y Biocatálisis, IBT-UNAM, AP 565-A, Cuernavaca, Morelos, México.
Mol Biol Evol. 2010 Jun;27(6):1449-59. doi: 10.1093/molbev/msq033. Epub 2010 Feb 1.
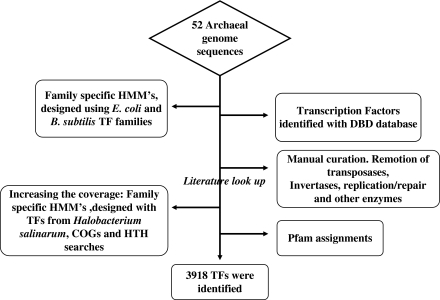
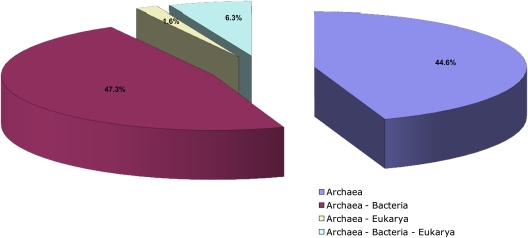
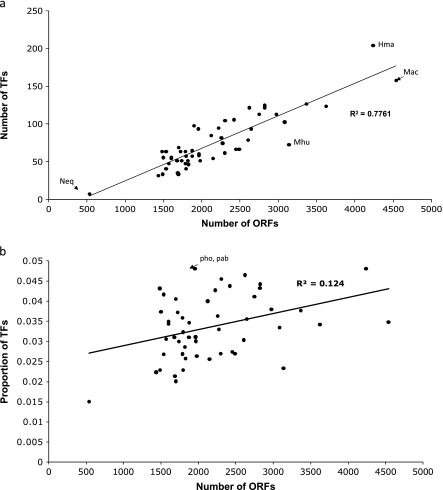
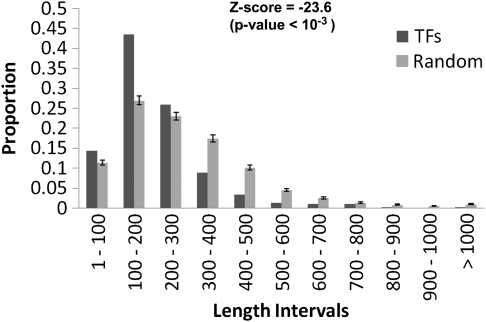
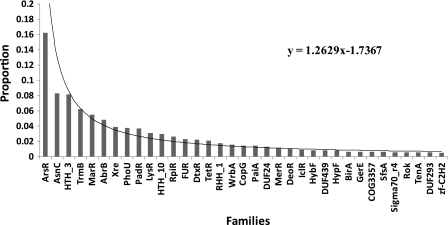
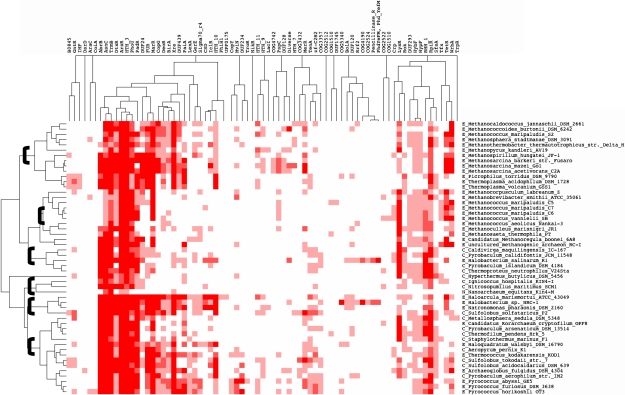
Archaea, which represent a large fraction of the phylogenetic diversity of organisms, are prokaryotes with eukaryote-like basal transcriptional machinery. This organization makes the study of their DNA-binding transcription factors (TFs) and their transcriptional regulatory networks particularly interesting. In addition, there are limited experimental data regarding their TFs. In this work, 3,918 TFs were identified and exhaustively analyzed in 52 archaeal genomes. TFs represented less than 5% of the gene products in all the studied species comparable with the number of TFs identified in parasites or intracellular pathogenic bacteria, suggesting a deficit in this class of proteins. A total of 75 families were identified, of which HTH_3, AsnC, TrmB, and ArsR families were universally and abundantly identified in all the archaeal genomes. We found that archaeal TFs are significantly small compared with other protein-coding genes in archaea as well as bacterial TFs, suggesting that a large fraction of these small-sized TFs could supply the probable deficit of TFs in archaea, by possibly forming different combinations of monomers similar to that observed in eukaryotic transcriptional machinery. Our results show that although the DNA-binding domains of archaeal TFs are similar to bacteria, there is an underrepresentation of ligand-binding domains in smaller TFs, which suggests that protein-protein interactions may act as mediators of regulatory feedback, indicating a chimera of bacterial and eukaryotic TFs' functionality. The analysis presented here contributes to the understanding of the details of transcriptional apparatus in archaea and provides a framework for the analysis of regulatory networks in these organisms.
古菌代表了生物系统发育多样性的很大一部分,是具有真核生物样基础转录机制的原核生物。这种组织使得研究它们的 DNA 结合转录因子(TFs)及其转录调控网络变得特别有趣。此外,关于它们的 TFs 的实验数据有限。在这项工作中,在 52 个古菌基因组中鉴定并详尽分析了 3918 个 TFs。TFs 在所有研究物种中的基因产物中所占比例不到 5%,与寄生虫或细胞内致病性细菌中鉴定的 TFs 数量相当,这表明这类蛋白质存在不足。共鉴定出 75 个家族,其中 HTH_3、AsnC、TrmB 和 ArsR 家族在所有古菌基因组中普遍且丰富地存在。我们发现,与古菌中的其他蛋白质编码基因以及细菌 TFs 相比,古菌 TFs 明显较小,这表明这些小尺寸 TFs 的很大一部分可能通过可能类似于真核转录机制中观察到的单体的不同组合来弥补古菌中 TFs 的可能不足。我们的结果表明,尽管古菌 TFs 的 DNA 结合结构域与细菌相似,但较小 TFs 中的配体结合结构域存在代表性不足,这表明蛋白质-蛋白质相互作用可能作为调控反馈的介质,表明细菌和真核 TFs 功能的嵌合体。这里呈现的分析有助于理解古菌转录装置的细节,并为这些生物体中调控网络的分析提供了框架。