Laboratory of Transcription Regulation, Department of Cell Biology, The Nencki Institute of Experimental Biology, Pasteur 3, 02-093 Warsaw, Poland.
BMC Bioinformatics. 2010 Feb 24;11:104. doi: 10.1186/1471-2105-11-104.
Affymetrix GeneChip microarrays are popular platforms for expression profiling in two types of studies: detection of differential expression computed by p-values of t-test and estimation of fold change between analyzed groups. There are many different preprocessing algorithms for summarizing Affymetrix data. The main goal of these methods is to remove effects of non-specific hybridization, and to optimally combine information from multiple probes annotated to the same transcript. The methods are benchmarked by comparison with reference methods, such as quantitative reverse-transcription PCR (qRT-PCR).
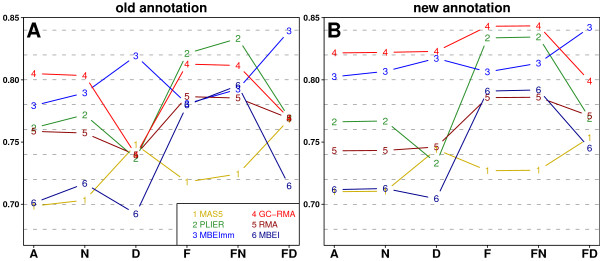
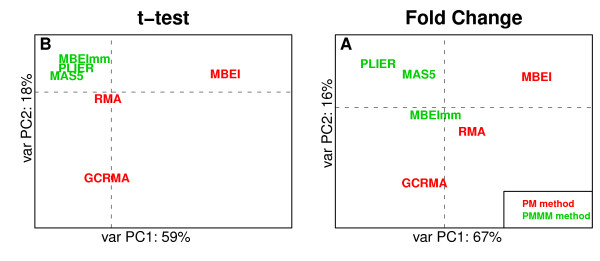
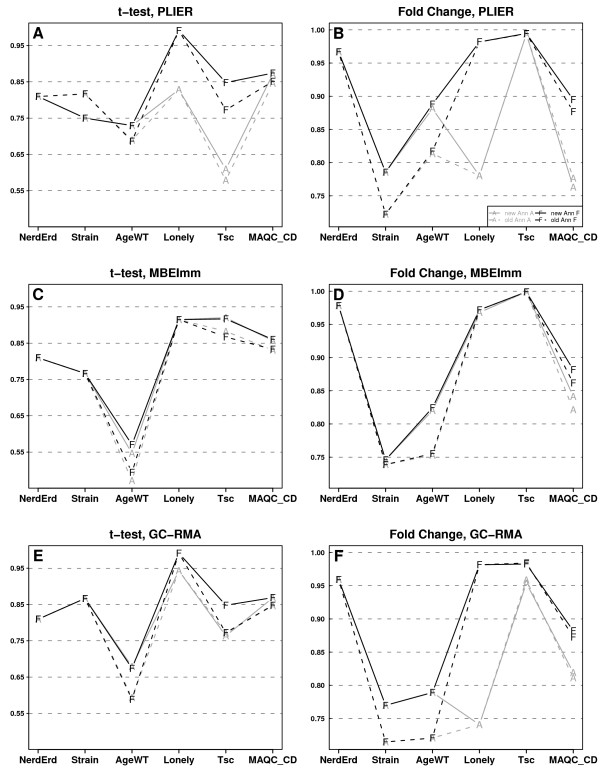
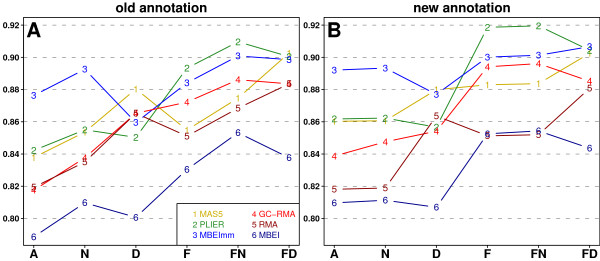
We present a comprehensive analysis of agreement between Affymetrix GeneChip and qRT-PCR results. We analyzed the influence of filtering by fraction Present calls introduced by J.N. McClintick and H.J. Edenberg (2006) and 2 mapping procedures: updated probe sets definitions proposed by Dai et al. (2005) and our "naive mapping" method. Because of evolution of genome sequence annotations since the time when microarrays were designed, we also studied the effect of the annotation release date. These comparisons were prepared for 6 popular preprocessing algorithms (MAS5, PLIER, RMA, GC-RMA, MBEI, and MBEImm) in the 2 above-mentioned types of studies. We used data sets from 6 independent biological experiments. As a measure of reproducibility of microarray and qRT-PCR values, we used linear and rank correlation coefficients.
We show that filtering by fraction Present calls increased correlations for all 6 preprocessing algorithms. We observed the difference in performance of PM-MM and PM-only methods: using MM probes increased correlations in fold change studies, but PM-only methods proved to perform better in detection of differential expression. We recommend using GC-RMA for detection of differential expression and PLIER for estimation of fold change. The use of the more recent annotation improves the results in both types of studies, encouraging re-analysis of old data.
Affymetrix GeneChip 微阵列是两种类型研究中表达谱分析的常用平台:通过 t 检验的 p 值检测差异表达,以及估计分析组之间的倍数变化。有许多不同的预处理算法可用于总结 Affymetrix 数据。这些方法的主要目标是消除非特异性杂交的影响,并优化组合注释到同一转录本的多个探针的信息。这些方法通过与参考方法(如定量逆转录 PCR(qRT-PCR))的比较进行基准测试。
我们对 Affymetrix GeneChip 和 qRT-PCR 结果之间的一致性进行了全面分析。我们分析了 J.N. McClintick 和 H.J. Edenberg(2006 年)提出的过滤分数 Present 调用和 2 种映射过程的影响:Dai 等人提出的更新探针集定义(2005 年)和我们的“原始映射”方法。由于微阵列设计时基因组序列注释的演变,我们还研究了注释发布日期的影响。这些比较是针对 6 种流行的预处理算法(MAS5、PLIER、RMA、GC-RMA、MBEI 和 MBEImm)在上述两种类型的研究中进行的。我们使用了来自 6 个独立生物学实验的数据。作为微阵列和 qRT-PCR 值重现性的衡量标准,我们使用了线性和等级相关系数。
我们表明,过滤分数 Present 调用增加了所有 6 种预处理算法的相关性。我们观察到 PM-MM 和 PM-Only 方法的性能差异:使用 MM 探针增加了倍数变化研究中的相关性,但 PM-Only 方法在检测差异表达方面表现更好。我们建议使用 GC-RMA 进行差异表达检测和 PLIER 进行倍数变化估计。在这两种类型的研究中,使用较新的注释可以提高结果,鼓励对旧数据进行重新分析。