Department of Medicine, University of Miami, 1120 NW 14th St, Suite 611, Miami, FL 33136, USA.
Bioinformatics. 2010 Apr 15;26(8):1043-9. doi: 10.1093/bioinformatics/btq097. Epub 2010 Mar 4.
Global expression patterns within cells are used for purposes ranging from the identification of disease biomarkers to basic understanding of cellular processes. Unfortunately, tissue samples used in cancer studies are usually composed of multiple cell types and the non-cancerous portions can significantly affect expression profiles. This severely limits the conclusions that can be made about the specificity of gene expression in the cell-type of interest. However, statistical analysis can be used to identify differentially expressed genes that are related to the biological question being studied.
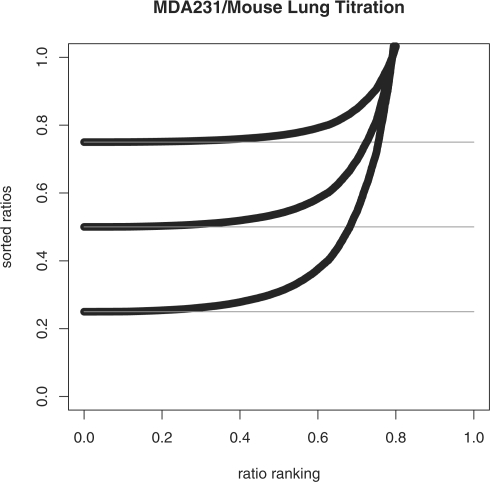
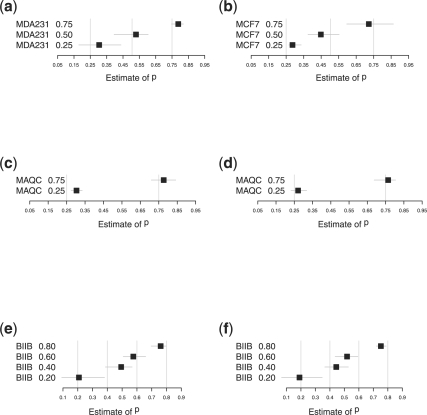
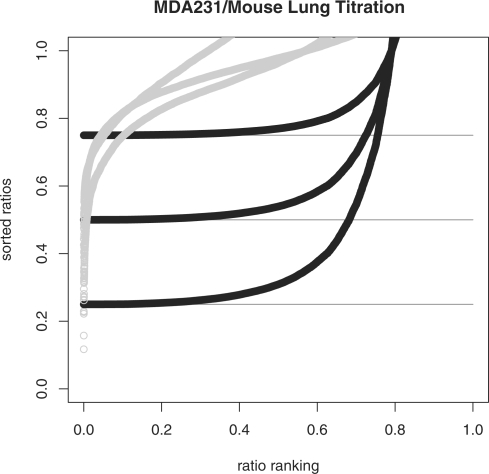
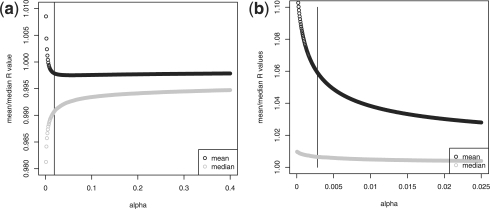
We propose a statistical approach to expression deconvolution from mixed tissue samples in which the proportion of each component cell type is unknown. Our method estimates the proportion of each component in a mixed tissue sample; this estimate can be used to provide estimates of gene expression from each component. We demonstrate our technique on xenograft samples from breast cancer research and publicly available experimental datasets found in the National Center for Biotechnology Information Gene Expression Omnibus repository.
R code (http://www.r-project.org/) for estimating sample proportions is freely available to non-commercial users and available at http://www.med.miami.edu/medicine/x2691.xml.
细胞内的全局表达模式可用于从疾病生物标志物的识别到细胞过程的基本理解等各种目的。不幸的是,癌症研究中使用的组织样本通常由多种细胞类型组成,非癌部分会显著影响表达谱。这严重限制了可以对感兴趣的细胞类型中的基因表达特异性做出的结论。但是,可以使用统计分析来识别与正在研究的生物学问题相关的差异表达基因。
我们提出了一种从混合组织样本中进行表达分解的统计方法,其中每个成分细胞类型的比例是未知的。我们的方法估计混合组织样本中每个成分的比例; 该估计值可用于提供来自每个成分的基因表达估计值。我们在乳腺癌研究的异种移植物样本和国家生物技术信息中心基因表达综合库中公开提供的可用实验数据集上证明了我们的技术。
用于估计样本比例的 R 代码(http://www.r-project.org/)可供非商业用户免费使用,并可在 http://www.med.miami.edu/medicine/x2691.xml 上获得。