Computer Science Department, Carnegie Mellon University, Pittsburgh PA 15213, USA.
Bioinformatics. 2010 Jun 15;26(12):i106-14. doi: 10.1093/bioinformatics/btq213.
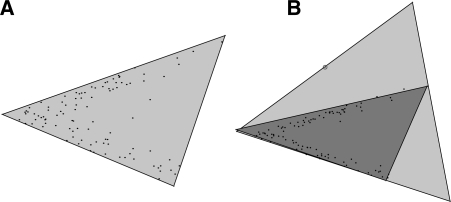
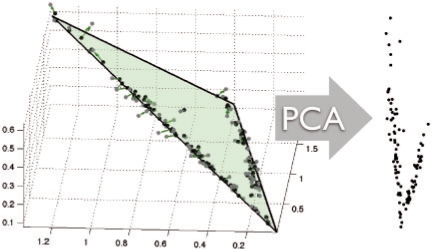
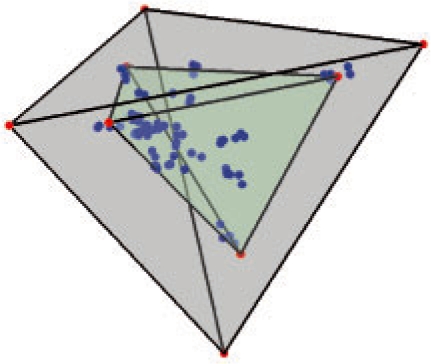
Tumorigenesis is an evolutionary process by which tumor cells acquire sequences of mutations leading to increased growth, invasiveness and eventually metastasis. It is hoped that by identifying the common patterns of mutations underlying major cancer sub-types, we can better understand the molecular basis of tumor development and identify new diagnostics and therapeutic targets. This goal has motivated several attempts to apply evolutionary tree reconstruction methods to assays of tumor state. Inference of tumor evolution is in principle aided by the fact that tumors are heterogeneous, retaining remnant populations of different stages along their development along with contaminating healthy cell populations. In practice, though, this heterogeneity complicates interpretation of tumor data because distinct cell types are conflated by common methods for assaying the tumor state. We previously proposed a method to computationally infer cell populations from measures of tumor-wide gene expression through a geometric interpretation of mixture type separation, but this approach deals poorly with noisy and outlier data.
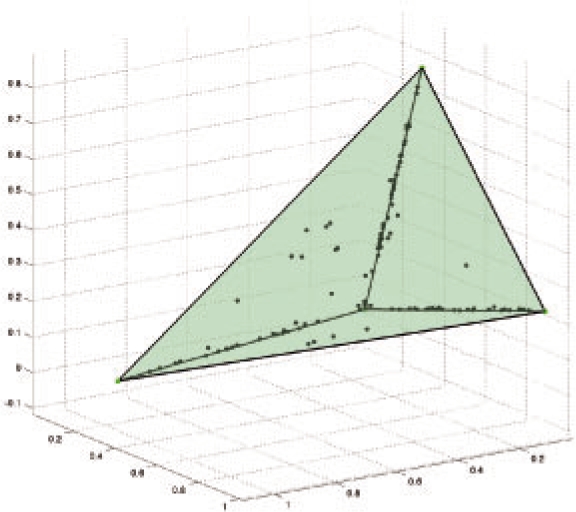
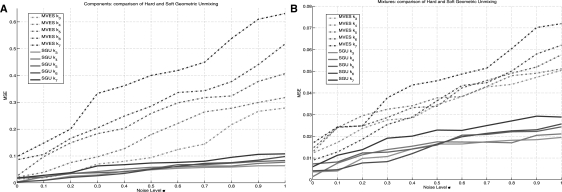
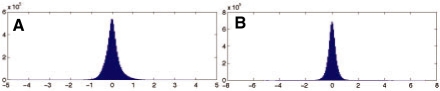
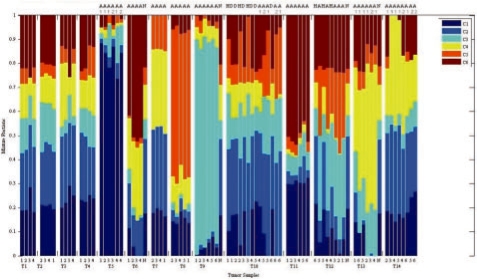
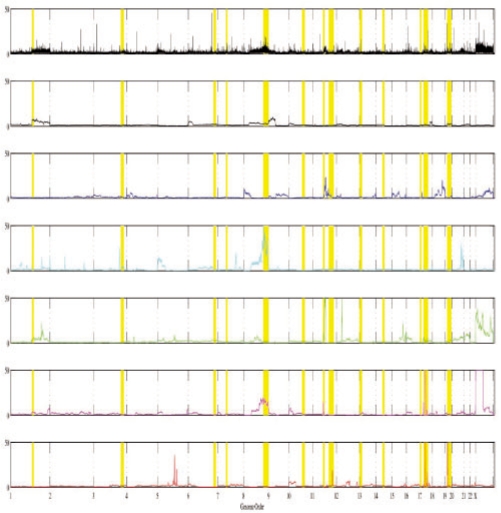
In the present work, we propose a new method to perform tumor mixture separation efficiently and robustly to an experimental error. The method builds on the prior geometric approach but uses a novel objective function allowing for robust fits that greatly reduces the sensitivity to noise and outliers. We further develop an efficient gradient optimization method to optimize this 'soft geometric unmixing' objective for measurements of tumor DNA copy numbers assessed by array comparative genomic hybridization (aCGH) data. We show, on a combination of semi-synthetic and real data, that the method yields fast and accurate separation of tumor states.
We have shown a novel objective function and optimization method for the robust separation of tumor sub-types from aCGH data and have shown that the method provides fast, accurate reconstruction of tumor states from mixed samples. Better solutions to this problem can be expected to improve our ability to accurately identify genetic abnormalities in primary tumor samples and to infer patterns of tumor evolution.
Supplementary data are available at Bioinformatics online.
肿瘤发生是一个进化过程,在此过程中,肿瘤细胞获得一系列突变序列,导致生长、侵袭性增加,最终转移。人们希望通过鉴定主要癌症亚型中潜在的常见突变模式,能够更好地理解肿瘤发生的分子基础,并确定新的诊断和治疗靶点。这一目标促使人们尝试将进化树重建方法应用于肿瘤状态的检测。从理论上讲,肿瘤的异质性有助于推断肿瘤的进化,因为肿瘤是异质的,在其发展过程中保留了不同阶段的残余种群,同时还混杂有健康细胞群体。然而,在实践中,这种异质性使肿瘤数据的解释变得复杂,因为不同的细胞类型通过用于检测肿瘤状态的常见方法而混淆在一起。我们之前提出了一种从肿瘤全基因表达测量中通过混合类型分离的几何解释来计算推断细胞群体的方法,但该方法对噪声和异常值数据的处理效果不佳。
在本工作中,我们提出了一种新的方法,可以有效地对实验误差进行稳健的肿瘤混合分离。该方法建立在先前的几何方法基础上,但使用了一个新的目标函数,允许稳健拟合,从而大大降低了对噪声和异常值的敏感性。我们进一步开发了一种有效的梯度优化方法,用于优化该“软几何解混”目标函数,以优化通过阵列比较基因组杂交(aCGH)数据评估的肿瘤 DNA 拷贝数的测量。我们结合半合成和真实数据进行了展示,表明该方法能够快速准确地分离肿瘤状态。
我们已经展示了一种新的目标函数和优化方法,用于从 aCGH 数据中稳健分离肿瘤亚型,并表明该方法能够从混合样本中快速、准确地重建肿瘤状态。对这个问题的更好解决方案有望提高我们准确识别原发性肿瘤样本中遗传异常的能力,并推断肿瘤进化的模式。
补充数据可在《生物信息学》在线获取。