Department of Infectious Disease Epidemiology, MRC Centre for Outbreak Analysis and Modelling, Imperial College Faculty of Medicine, London, UK.
Heredity (Edinb). 2011 Feb;106(2):383-90. doi: 10.1038/hdy.2010.78. Epub 2010 Jun 16.
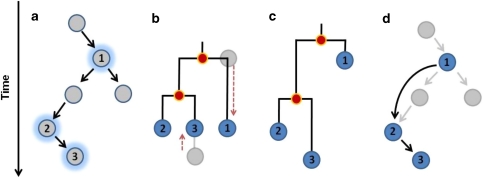
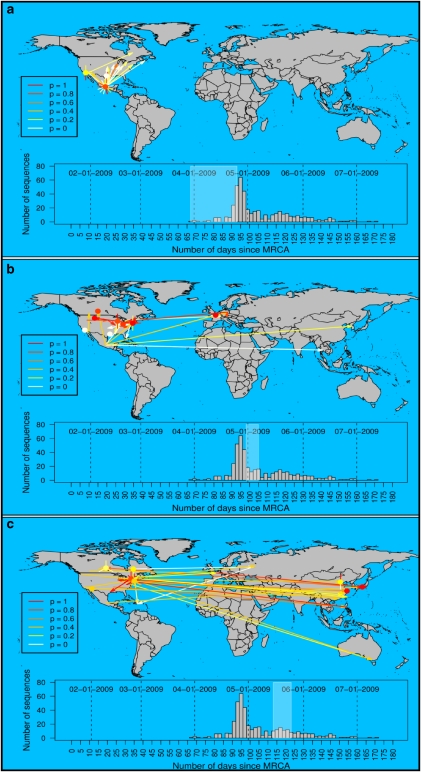
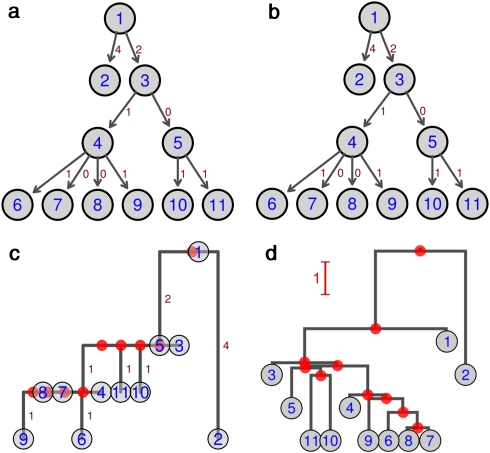
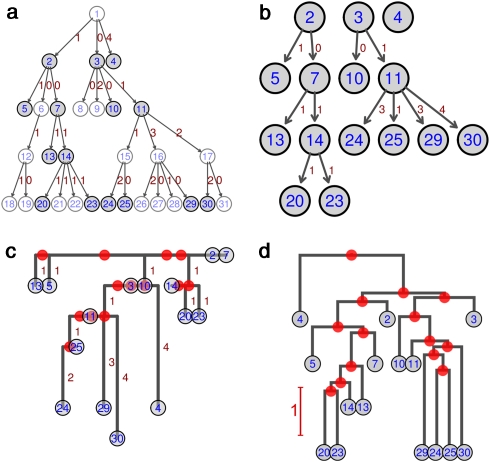
Epidemiology and public health planning will increasingly rely on the analysis of genetic sequence data. In particular, genetic data coupled with dates and locations of sampled isolates can be used to reconstruct the spatiotemporal dynamics of pathogens during outbreaks. Thus far, phylogenetic methods have been used to tackle this issue. Although these approaches have proved useful for informing on the spread of pathogens, they do not aim at directly reconstructing the underlying transmission tree. Instead, phylogenetic models infer most recent common ancestors between pairs of isolates, which can be inadequate for densely sampled recent outbreaks, where the sample includes ancestral and descendent isolates. In this paper, we introduce a novel method based on a graph approach to reconstruct transmission trees directly from genetic data. Using simulated data, we show that our approach can efficiently reconstruct genealogies of isolates in situations where classical phylogenetic approaches fail to do so. We then illustrate our method by analyzing data from the early stages of the swine-origin A/H1N1 influenza pandemic. Using 433 isolates sequenced at both the hemagglutinin and neuraminidase genes, we reconstruct the likely history of the worldwide spread of this new influenza strain. The presented methodology opens new perspectives for the analysis of genetic data in the context of disease outbreaks.
流行病学和公共卫生规划将越来越依赖于对基因序列数据的分析。特别是,将基因数据与采样分离株的日期和位置相结合,可以用于重建传染病爆发期间病原体的时空动态。到目前为止,已经使用系统发育学方法来解决这个问题。尽管这些方法已被证明对了解病原体的传播很有用,但它们并不是旨在直接重建潜在的传播树。相反,系统发育模型推断分离株之间的最近共同祖先,对于最近密集采样的爆发情况,其中样本包括祖先和后代分离株,这种方法可能不够充分。在本文中,我们引入了一种基于图方法的新方法,可直接从遗传数据中重建传播树。使用模拟数据,我们表明,在经典系统发育方法无法进行的情况下,我们的方法可以有效地重建分离株的系统发育。然后,我们通过分析猪源 A/H1N1 流感大流行早期的数据来说明我们的方法。使用在血凝素和神经氨酸酶基因上均进行测序的 433 个分离株,我们重建了这种新流感病毒株在全球传播的可能历史。所提出的方法为疾病爆发背景下的遗传数据分析开辟了新的视角。