Center for Computational Immunology, Duke University Medical Center, Durham, NC, USA.
BMC Genomics. 2010 Jul 21;11:444. doi: 10.1186/1471-2164-11-444.
The rate of emergence of human pathogens is steadily increasing; most of these novel agents originate in wildlife. Bats, remarkably, are the natural reservoirs of many of the most pathogenic viruses in humans. There are two bat genome projects currently underway, a circumstance that promises to speed the discovery host factors important in the coevolution of bats with their viruses. These genomes, however, are not yet assembled and one of them will provide only low coverage, making the inference of most genes of immunological interest error-prone. Many more wildlife genome projects are underway and intend to provide only shallow coverage.
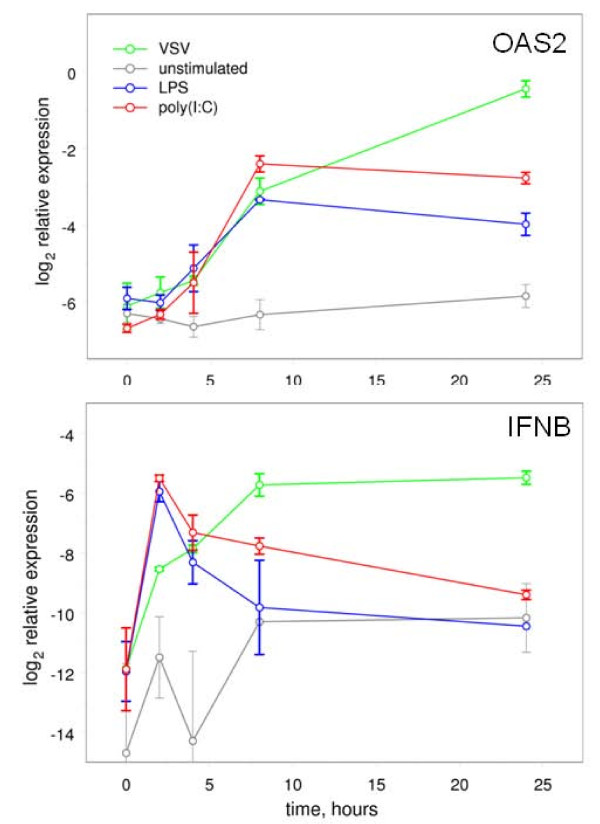
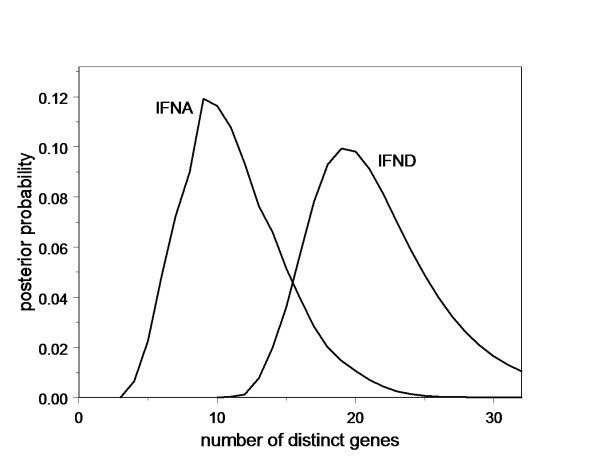
We have developed a statistical method for the assembly of gene families from partial genomes. The method takes full advantage of the quality scores generated by base-calling software, incorporating them into a complete probabilistic error model, to overcome the limitation inherent in the inference of gene family members from partial sequence information. We validated the method by inferring the human IFNA genes from the genome trace archives, and used it to infer 61 type-I interferon genes, and single type-II interferon genes in the bats Pteropus vampyrus and Myotis lucifugus. We confirmed our inferences by direct cloning and sequencing of IFNA, IFNB, IFND, and IFNK in P. vampyrus, and by demonstrating transcription of some of the inferred genes by known interferon-inducing stimuli.
The statistical trace assembler described here provides a reliable method for extracting information from the many available and forthcoming partial or shallow genome sequencing projects, thereby facilitating the study of a wider variety of organisms with ecological and biomedical significance to humans than would otherwise be possible.
人类病原体的出现率正在稳步上升;这些新病原体大多源自野生动物。值得注意的是,蝙蝠是许多对人类最具致病性病毒的天然宿主。目前有两个蝙蝠基因组项目正在进行中,这一情况有望加速发现宿主因子在蝙蝠与其病毒的共同进化中的重要作用。然而,这些基因组尚未组装,其中一个基因组的覆盖度很低,这使得推断大多数具有免疫意义的基因容易出错。还有更多的野生动物基因组项目正在进行中,它们打算只提供浅层覆盖度。
我们开发了一种从部分基因组组装基因家族的统计方法。该方法充分利用碱基调用软件生成的质量分数,将其纳入完整的概率错误模型中,克服了从部分序列信息推断基因家族成员所固有的局限性。我们通过从基因组轨迹档案中推断人类 IFNA 基因来验证该方法,并使用该方法推断了蝙蝠 Pteropus vampyrus 和 Myotis lucifugus 中的 61 种 I 型干扰素基因和单个 II 型干扰素基因。我们通过直接克隆和测序 P. vampyrus 中的 IFNA、IFNB、IFND 和 IFNK 以及证明一些推断基因对已知干扰素诱导刺激物的转录,证实了我们的推断。
这里描述的统计轨迹组装器提供了一种可靠的方法,可以从许多现有的和即将到来的部分或浅层基因组测序项目中提取信息,从而促进对具有生态和医学意义的更广泛种类的生物体的研究,这些生物体对人类的意义比其他方法所能实现的要大。