Center for the Study of Systems Biology, School of Biology, Georgia Institute of Technology, Atlanta, Georgia 30318, USA.
J Chem Inf Model. 2010 Oct 25;50(10):1839-54. doi: 10.1021/ci100235n.
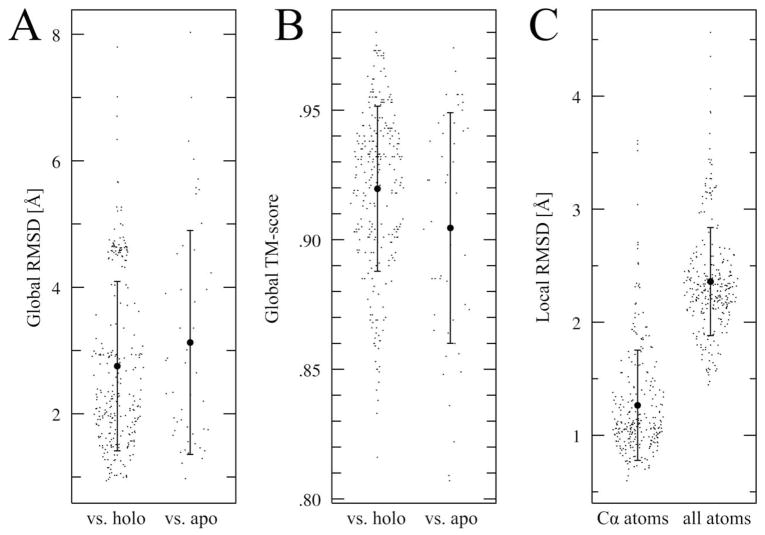
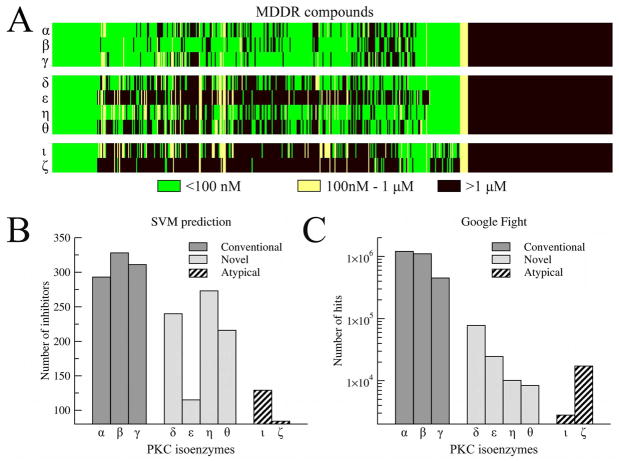
The growing interest in the identification of kinase inhibitors, promising therapeutics in the treatment of many diseases, has created a demand for the structural characterization of the entire human kinome. At the outset of the drug development process, the lead-finding stage, approaches that enrich the screening library with bioactive compounds are needed. Here, protein structure based methods can play an important role, but despite structural genomics efforts, it is unlikely that the three-dimensional structures of the entire kinome will be available soon. Therefore, at the proteome level, structure-based approaches must rely on predicted models, with a key issue being their utility in virtual ligand screening. In this study, we employ the recently developed FINDSITE/Q-Dock ligand homology modeling approach, which is well-suited for proteome-scale applications using predicted structures, to provide extensive structural and functional characterization of the human kinome. Specifically, we construct structure models for the human kinome; these are subsequently subject to virtual screening against a library of more than 2 million compounds. To rank the compounds, we employ a hierarchical approach that combines ligand- and structure-based filters. Modeling accuracy is carefully validated using available experimental data with particularly encouraging results found for the ability to identify, without prior knowledge, specific kinase inhibitors. More generally, the modeling procedure results in a large number of predicted molecular interactions between kinases and small ligands that should be of practical use in the development of novel inhibitors. The data set is freely available to the academic community via a user-friendly Web interface at http://cssb.biology.gatech.edu/kinomelhm/ as well as at the ZINC Web site ( http://zinc.docking.org/applications/2010Apr/Brylinski-2010.tar.gz ).
人们对鉴定激酶抑制剂越来越感兴趣,而这类抑制剂在治疗多种疾病方面具有广阔的应用前景,这也促使人们对整个人类激酶组进行结构特征分析。在药物开发过程的起始阶段,即先导化合物发现阶段,需要采用能够使生物活性化合物在筛选库中得到富集的方法。在这一阶段,基于蛋白质结构的方法可以发挥重要作用,但是尽管已经开展了结构基因组学方面的研究工作,要想在近期内获得整个激酶组的三维结构仍然不太可能。因此,在蛋白质组水平上,基于结构的方法必须依赖预测模型,而关键问题是这些模型在虚拟配体筛选中的实用性。在本研究中,我们采用了最近开发的 FINDSITE/Q-Dock 配体同源建模方法,该方法非常适合于使用预测结构进行大规模蛋白质组应用,从而对人类激酶组进行广泛的结构和功能分析。具体而言,我们构建了人类激酶组的结构模型,然后对这些模型进行了虚拟筛选,筛选对象是一个包含 200 多万种化合物的文库。为了对化合物进行排序,我们采用了一种层次化的方法,该方法结合了配体和结构两种过滤方式。我们使用现有的实验数据仔细验证了建模的准确性,结果特别令人鼓舞,因为我们能够在没有先验知识的情况下识别出特定的激酶抑制剂。更普遍地说,建模过程产生了大量激酶与小分子配体之间的预测分子相互作用,这些相互作用数据应该能够在新型抑制剂的开发中得到实际应用。数据集通过用户友好的 Web 界面(http://cssb.biology.gatech.edu/kinomelhm/)和 ZINC 网站(http://zinc.docking.org/applications/2010Apr/Brylinski-2010.tar.gz)免费提供给学术界。