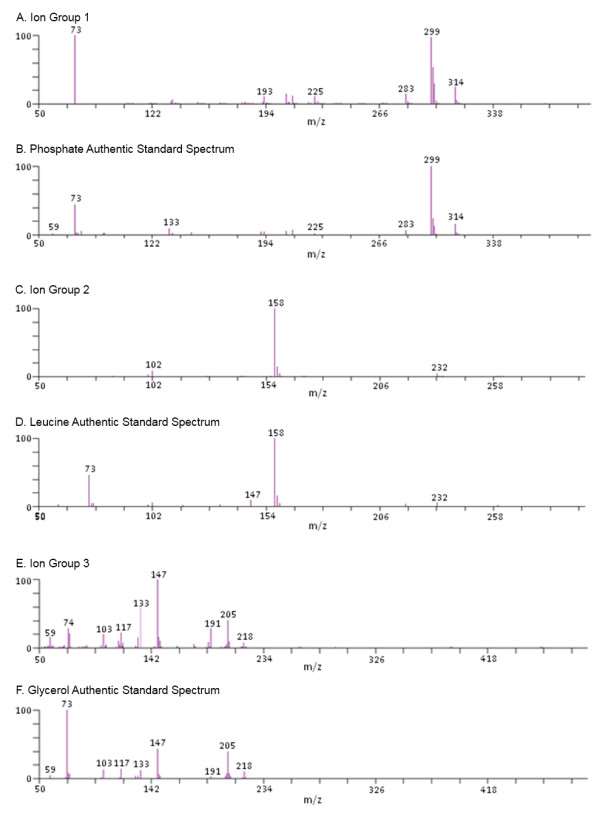
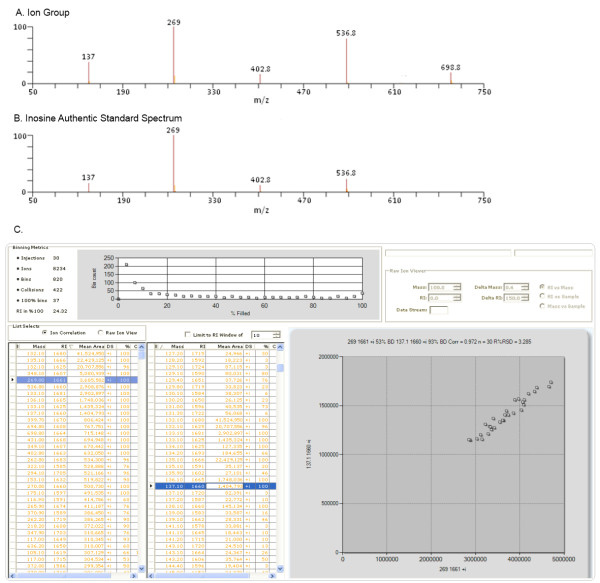
Metabolon, Inc,, 800 Capitola Drive, Suite 1, Durham, NC 27713, USA.
J Cheminform. 2010 Oct 18;2(1):9. doi: 10.1186/1758-2946-2-9.
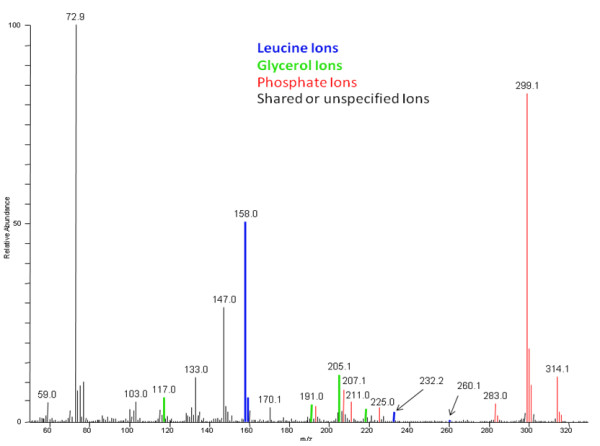
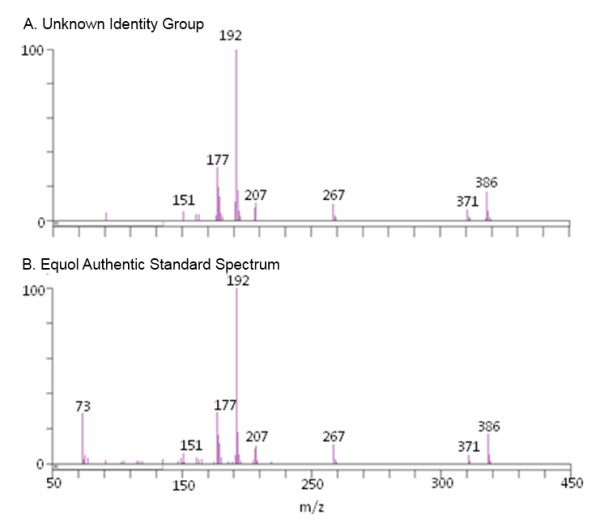
Metabolomics experiments involve generating and comparing small molecule (metabolite) profiles from complex mixture samples to identify those metabolites that are modulated in altered states (e.g., disease, drug treatment, toxin exposure). One non-targeted metabolomics approach attempts to identify and interrogate all small molecules in a sample using GC or LC separation followed by MS or MSn detection. Analysis of the resulting large, multifaceted data sets to rapidly and accurately identify the metabolites is a challenging task that relies on the availability of chemical libraries of metabolite spectral signatures. A method for analyzing spectrometry data to identify and Quantify Individual Components in a Sample, (QUICS), enables generation of chemical library entries from known standards and, importantly, from unknown metabolites present in experimental samples but without a corresponding library entry. This method accounts for all ions in a sample spectrum, performs library matches, and allows review of the data to quality check library entries. The QUICS method identifies ions related to any given metabolite by correlating ion data across the complete set of experimental samples, thus revealing subtle spectral trends that may not be evident when viewing individual samples and are likely to be indicative of the presence of one or more otherwise obscured metabolites.
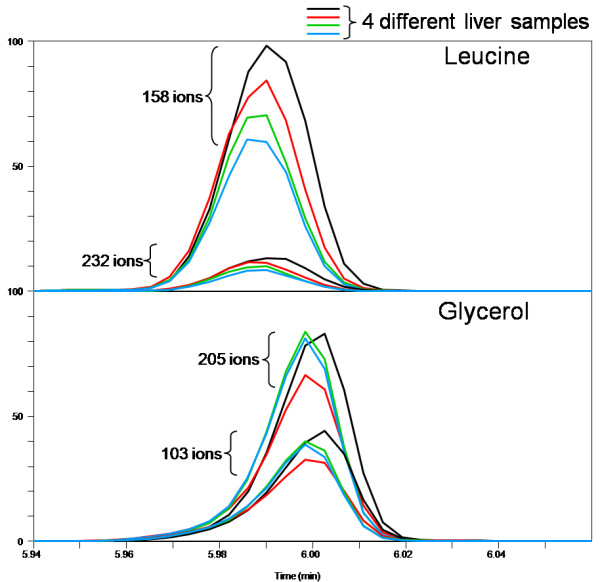
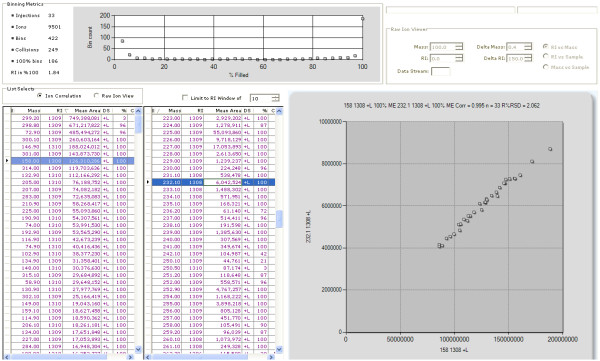
LC-MS/MS or GC-MS data from 33 liver samples were analyzed simultaneously which exploited the inherent biological diversity of the samples and the largely non-covariant chemical nature of the metabolites when viewed over multiple samples. Ions were partitioned by both retention time (RT) and covariance which grouped ions from a single common underlying metabolite. This approach benefitted from using mass, time and intensity data in aggregate over the entire sample set to reject outliers and noise thereby producing higher quality chemical identities. The aggregated data was matched to reference chemical libraries to aid in identifying the ion set as a known metabolite or as a new unknown biochemical to be added to the library.
The QUICS methodology enabled rapid, in-depth evaluation of all possible metabolites (known and unknown) within a set of samples to identify the metabolites and, for those that did not have an entry in the reference library, to create a library entry to identify that metabolite in future studies.
代谢组学实验涉及生成和比较复杂混合物样本中的小分子(代谢物)谱,以鉴定在改变的状态下(例如,疾病、药物治疗、毒素暴露)被调节的代谢物。一种非靶向代谢组学方法试图使用 GC 或 LC 分离,然后使用 MS 或 MSn 检测来识别和检测样品中的所有小分子。分析由此产生的大型、多方面的数据以快速准确地识别代谢物是一项具有挑战性的任务,这依赖于代谢物光谱特征化学库的可用性。一种用于分析光谱数据以鉴定和定量样品中单个成分的方法(QUICS),能够从已知标准和重要的从实验样品中存在的未知代谢物生成化学库条目,但没有相应的库条目。该方法考虑了样品光谱中的所有离子,进行了库匹配,并允许对数据进行质量检查以检查库条目。QUICS 方法通过跨整个实验样品集相关离子数据来识别与任何给定代谢物相关的离子,从而揭示出在查看单个样品时可能不明显的细微光谱趋势,并且可能表明存在一个或多个否则被掩盖的代谢物。
同时分析了 33 个肝样本的 LC-MS/MS 或 GC-MS 数据,这利用了样本的固有生物学多样性以及在多个样本中观察到的代谢物的主要非协变化学性质。离子通过保留时间(RT)和协方差进行分区,这将来自单个共同基础代谢物的离子分组。这种方法受益于在整个样品集中使用质量、时间和强度数据的集合来拒绝异常值和噪声,从而产生更高质量的化学身份。聚合数据与参考化学库匹配,以帮助识别离子集为已知代谢物或添加到库中的新未知生化物质。
QUICS 方法学能够快速、深入地评估一组样本中所有可能的代谢物(已知和未知),以鉴定代谢物,对于那些在参考库中没有条目的代谢物,创建一个库条目以在未来的研究中识别该代谢物。