Institute of Evolutionary Biology, CSIC-UPF, Passeig Maritim de la Barceloneta 37, 08003 Barcelona, Spain.
BMC Evol Biol. 2010 Nov 30;10:369. doi: 10.1186/1471-2148-10-369.
Multilocus phylogenies can be used to infer the species tree of a group of closely related species. In species trees, the nodes represent the actual separation between species, thus providing essential information about their evolutionary history. In addition, multilocus phylogenies can help in analyses of species delimitation, gene flow and genetic differentiation within species. However, few adequate markers are available for such studies.
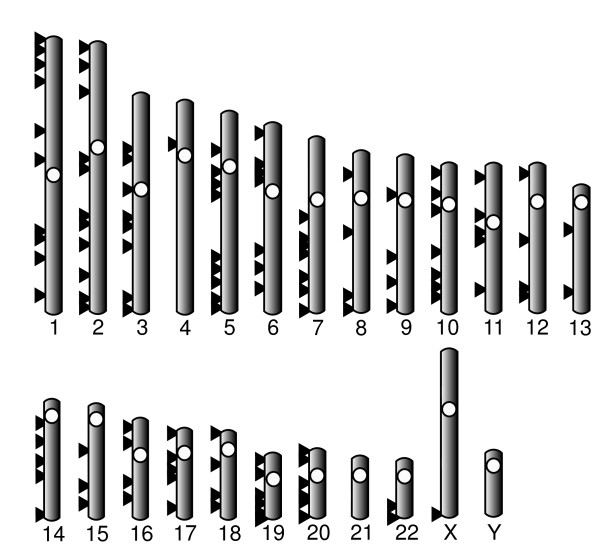
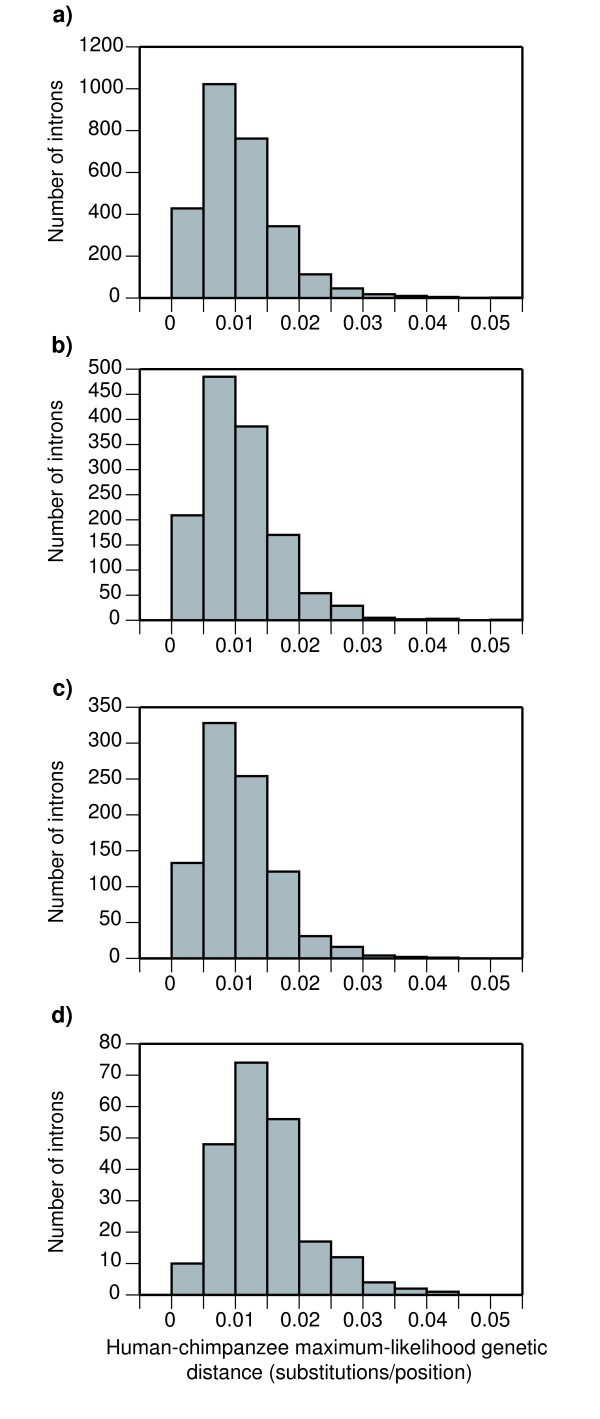
In order to develop nuclear markers that can be useful in multilocus studies of mammals, we analyzed the mammalian genomes of human, chimpanzee, macaque, dog and cow. Rodents were excluded due to their unusual genomic features. Introns were extracted from the mammalian genomes because of their greater genetic variability and ease of amplification from the flanking exons. To an initial set of more than 10,000 one-to-one orthologous introns we applied several filters to select introns that belong to single-copy genes, show neutral evolutionary rates and have an adequate length for their amplification. This analysis led to a final list of 224 intron markers randomly distributed along the genome. To experimentally test their validity, we amplified twelve of these introns in a panel of six mammalian species. The result was that seven of these introns gave rise to a PCR band of the expected size in all species. In addition, we sequenced these bands and analyzed the accumulation of substitutions in these introns in five pairs of closely related species. The results showed that the estimated genetic distances in the five species pairs was quite variable among introns and that this divergence cannot be directly predicted from the overall intron divergence in mammals.
We have designed a new set of 224 nuclear introns with optimal features for the phylogeny of closely related mammalian species. A large proportion of the introns tested experimentally showed a perfect amplification and enough variability in most species, indicating that this marker set can be very helpful in multilocus phylogenetics of mammals. Due to the lower variability and stronger stochasticity of nuclear markers with respect to mitochondrial genes, studies should be designed to make use of several markers like the ones designed here.
多位点系统发育树可用于推断一组密切相关物种的种系发生树。在种系发生树中,节点代表物种之间的实际分离,从而提供有关其进化历史的重要信息。此外,多位点系统发育树有助于分析物种界限、基因流和物种内的遗传分化。然而,很少有足够的标记可用于此类研究。
为了开发可用于哺乳动物多位点研究的核标记,我们分析了人类、黑猩猩、猕猴、狗和牛的哺乳动物基因组。由于其异常的基因组特征,排除了啮齿动物。选择从哺乳动物基因组中提取内含子,因为它们具有更大的遗传变异性,并且从侧翼外显子扩增更容易。对于最初的超过 10000 个一对一的直系同源内含子,我们应用了几个过滤器来选择属于单拷贝基因的内含子、显示中性进化率并具有足够长度用于扩增的内含子。该分析导致了 224 个随机分布在基因组中的内含子标记的最终列表。为了实验验证其有效性,我们在 6 种哺乳动物的小组中扩增了这 12 个内含子。结果是,这 7 个内含子在所有物种中产生了预期大小的 PCR 带。此外,我们对这些带进行了测序,并分析了在 5 对密切相关的物种中这些内含子的替换积累。结果表明,在 5 个物种对中,估计的遗传距离在不同内含子之间差异很大,并且这种分歧不能直接从哺乳动物总体内含子分歧中预测。
我们设计了一组新的 224 个核内含子,这些内含子具有密切相关哺乳动物物种系统发育的最佳特征。在实验中测试的大部分内含子在大多数物种中显示出完美的扩增和足够的可变性,表明该标记集在哺乳动物多位点系统发育学中非常有用。由于相对于线粒体基因,核标记的变异性和随机性较低,因此应该设计研究来利用像这里设计的那样的多个标记。