Department of Molecular Biology, Radboud University, Nijmegen Center for Molecular Life Sciences, Nijmegen, The Netherlands.
PLoS Pathog. 2010 Dec 16;6(12):e1001223. doi: 10.1371/journal.ppat.1001223.
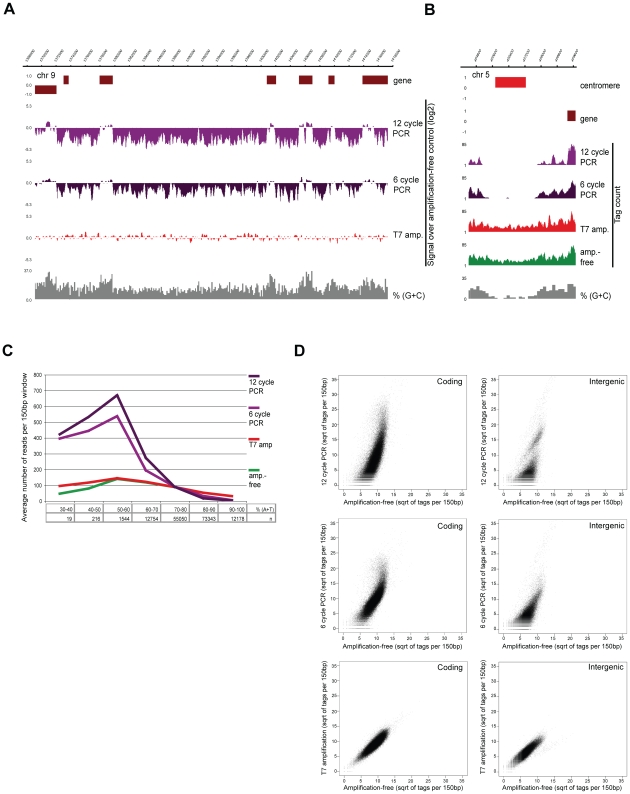
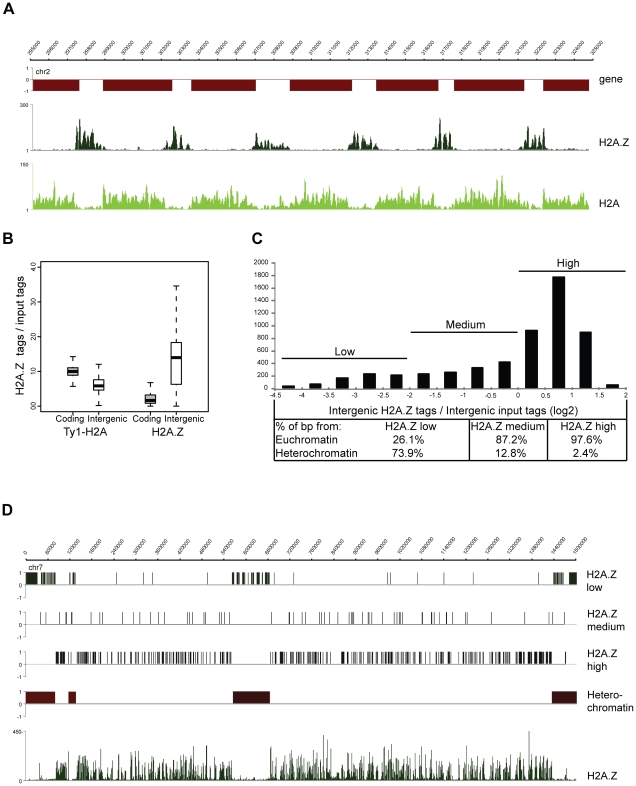
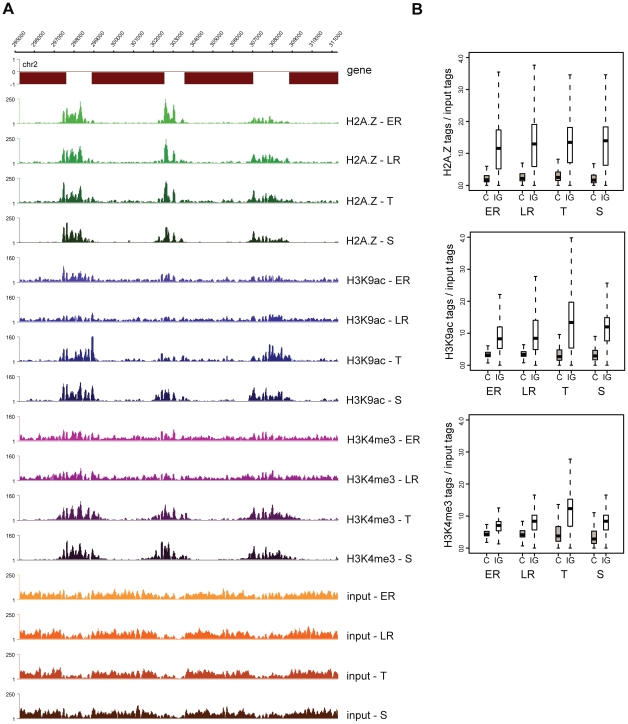
Epigenetic regulatory mechanisms and their enzymes are promising targets for malaria therapeutic intervention; however, the epigenetic component of gene expression in P. falciparum is poorly understood. Dynamic or stable association of epigenetic marks with genomic features provides important clues about their function and helps to understand how histone variants/modifications are used for indexing the Plasmodium epigenome. We describe a novel, linear amplification method for next-generation sequencing (NGS) that allows unbiased analysis of the extremely AT-rich Plasmodium genome. We used this method for high resolution, genome-wide analysis of a histone H2A variant, H2A.Z and two histone H3 marks throughout parasite intraerythrocytic development. Unlike in other organisms, H2A.Z is a constant, ubiquitous feature of euchromatic intergenic regions throughout the intraerythrocytic cycle. The almost perfect colocalisation of H2A.Z with H3K9ac and H3K4me3 suggests that these marks are preferentially deposited on H2A.Z-containing nucleosomes. By performing RNA-seq on 8 time-points, we show that acetylation of H3K9 at promoter regions correlates very well with the transcriptional status whereas H3K4me3 appears to have stage-specific regulation, being low at early stages, peaking at trophozoite stage, but does not closely follow changes in gene expression. Our improved NGS library preparation procedure provides a foundation to exploit the malaria epigenome in detail. Furthermore, our findings place H2A.Z at the cradle of P. falciparum epigenetic regulation by stably defining intergenic regions and providing a platform for dynamic assembly of epigenetic and other transcription related complexes.
表观遗传调控机制及其酶类是疟疾治疗干预的有前途的靶点;然而,疟原虫中基因表达的表观遗传成分还知之甚少。表观遗传标记与基因组特征的动态或稳定关联为它们的功能提供了重要线索,并有助于了解组蛋白变体/修饰如何用于标记疟原虫的表观基因组。我们描述了一种新的、用于下一代测序(NGS)的线性扩增方法,该方法允许对极度富含 AT 的疟原虫基因组进行无偏分析。我们使用该方法对组蛋白 H2A 变体 H2A.Z 和两种组蛋白 H3 标记在寄生虫红细胞内发育过程中的全基因组进行高分辨率分析。与其他生物体不同的是,H2A.Z 是整个红细胞内周期中常染色质基因间区的恒定、普遍特征。H2A.Z 与 H3K9ac 和 H3K4me3 的近乎完美共定位表明,这些标记优先沉积在含有 H2A.Z 的核小体上。通过在 8 个时间点进行 RNA-seq,我们表明 H3K9 的乙酰化在启动子区域与转录状态非常相关,而 H3K4me3 似乎具有阶段特异性调节,在早期阶段较低,在滋养体阶段达到峰值,但并不密切跟随基因表达的变化。我们改进的 NGS 文库制备程序为详细研究疟原虫的表观基因组提供了基础。此外,我们的发现将 H2A.Z 置于疟原虫表观遗传调控的摇篮中,通过稳定定义基因间区并为动态组装表观遗传和其他转录相关复合物提供平台。