National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing, China.
PLoS One. 2011 Feb 18;6(2):e17215. doi: 10.1371/journal.pone.0017215.
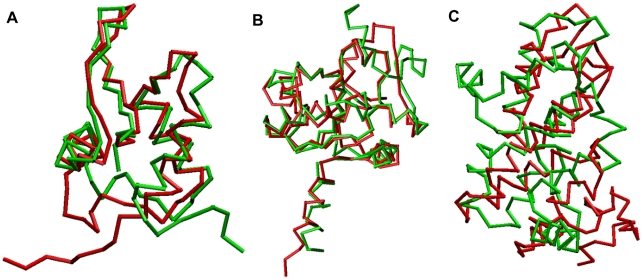
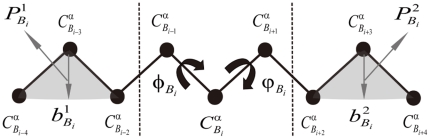
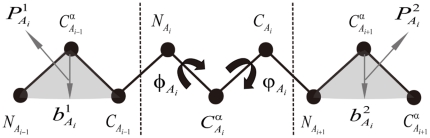
Fold recognition, or threading, is a popular protein structure modeling approach that uses known structure templates to build structures for those of unknown. The key to the success of fold recognition methods lies in the proper integration of sequence, physiochemical and structural information. Here we introduce another type of information, local structural preference potentials of 3-residue and 9-residue fragments, for fold recognition. By combining the two local structural preference potentials with the widely used sequence profile, secondary structure information and hydrophobic score, we have developed a new threading method called FR-t5 (fold recognition by use of 5 terms). In benchmark testings, we have found the consideration of local structural preference potentials in FR-t5 not only greatly enhances the alignment accuracy and recognition sensitivity, but also significantly improves the quality of prediction models.
折叠识别,或穿线,是一种流行的蛋白质结构建模方法,它使用已知的结构模板来构建未知结构的结构。折叠识别方法成功的关键在于正确整合序列、物理化学和结构信息。在这里,我们引入另一种信息,即 3 残基和 9 残基片段的局部结构偏好势,用于折叠识别。通过将这两种局部结构偏好势与广泛使用的序列轮廓、二级结构信息和疏水性得分相结合,我们开发了一种新的穿线方法,称为 FR-t5(使用 5 个术语进行折叠识别)。在基准测试中,我们发现 FR-t5 中考虑局部结构偏好势不仅大大提高了对齐精度和识别灵敏度,而且还显著提高了预测模型的质量。