Laboratory for Conservation and Utilization of Bio-resource, Yunnan University, Kunming, 650091, PR, China.
BMC Evol Biol. 2011 Apr 10;11:92. doi: 10.1186/1471-2148-11-92.
Mustelidae, as the largest and most-diverse family of order Carnivora, comprises eight subfamilies. Phylogenetic relationships among these Mustelidae subfamilies remain argumentative subjects in recent years. One of the main reasons is that the mustelids represent a typical example of rapid evolutionary radiation and recent speciation event. Prior investigation has been concentrated on the application of different mitochondrial (mt) sequence and nuclear protein-coding data, herein we employ 17 nuclear non-coding loci (>15 kb), in conjunction with mt complete genome data (>16 kb), to clarify these enigmatic problems.
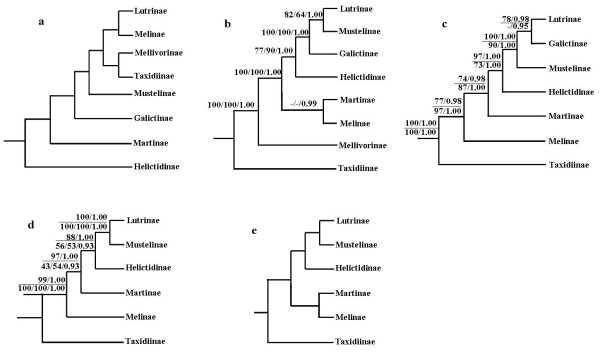
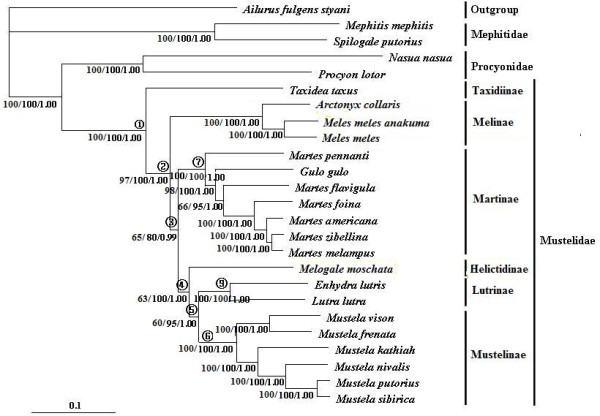
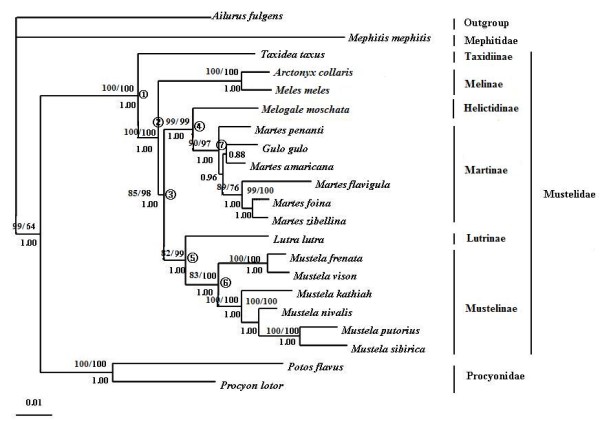
The combined nuclear intron and mt genome analyses both robustly support that Taxidiinae diverged first, followed by Melinae. Lutrinae and Mustelinae are grouped together in all analyses with strong supports. The position of Helictidinae, however, is enigmatic because the mt genome analysis places it to the clade uniting Lutrinae and Mustelinae, whereas the nuclear intron analysis favors a novel view supporting a closer relationship of Helictidinae to Martinae. This finding emphasizes a need to add more data and include more taxa to resolve this problem. In addition, the molecular dating provides insights into the time scale of the origin and diversification of the Mustelidae subfamilies. Finally, the phylogenetic performances and limits of nuclear introns and mt genes are discussed in the context of Mustelidae phylogeny.
Our study not only brings new perspectives on the previously obscured phylogenetic relationships among Mustelidae subfamilies, but also provides another example demonstrating the effectiveness of nuclear non-coding loci for reconstructing evolutionary histories in a group that has undergone rapid bursts of speciation.
鼬科是食肉目最大、最多样化的科,包括 8 个亚科。近年来,鼬科亚科之间的系统发育关系仍是争议的主题。其中一个主要原因是鼬科是快速进化辐射和近期物种形成事件的典型代表。先前的研究主要集中在不同的线粒体(mt)序列和核蛋白编码数据的应用上,在这里我们采用了 17 个核非编码基因座(>15kb),结合 mt 完整基因组数据(>16kb),以阐明这些神秘的问题。
联合核内含子和 mt 基因组分析都强有力地支持了獾亚科首先分化,其次是貂亚科。所有分析都将水獭亚科和鼬亚科聚在一起,支持力度很强。獾亚科的位置则很神秘,因为 mt 基因组分析将其置于与水獭亚科和鼬亚科联合的分支上,而核内含子分析则支持一个新颖的观点,即獾亚科与貂亚科关系更密切。这一发现强调需要增加更多的数据并纳入更多的分类单元来解决这个问题。此外,分子定年为鼬科亚科的起源和多样化时间尺度提供了新的视角。最后,在鼬科的系统发育中讨论了核内含子和 mt 基因的系统发育表现和局限性。
我们的研究不仅为鼬科亚科之间以前模糊的系统发育关系带来了新的视角,而且还提供了另一个例子,证明了核非编码基因座在重建经历快速物种形成爆发的群体进化历史方面的有效性。