Bucher Denis, Pierce Levi C T, McCammon J Andrew, Markwick Phineus R L
Department of Chemistry and Biochemistry, University of California , San Diego, 9500 Gilman Drive, La Jolla, California 92093- 0365, United States.
J Chem Theory Comput. 2011 Apr 12;7(4):890-897. doi: 10.1021/ct100605v. Epub 2011 Mar 4.
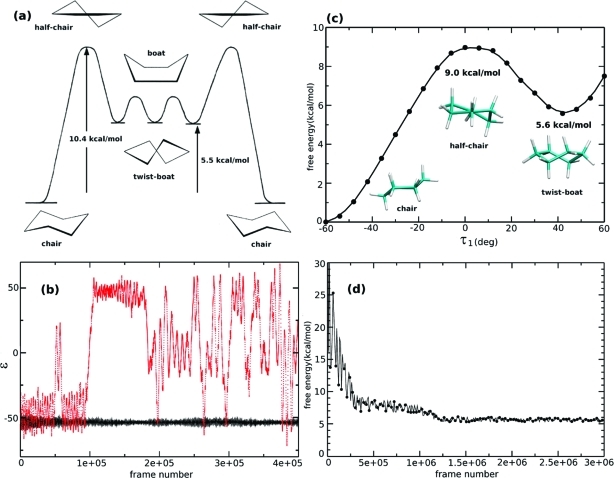
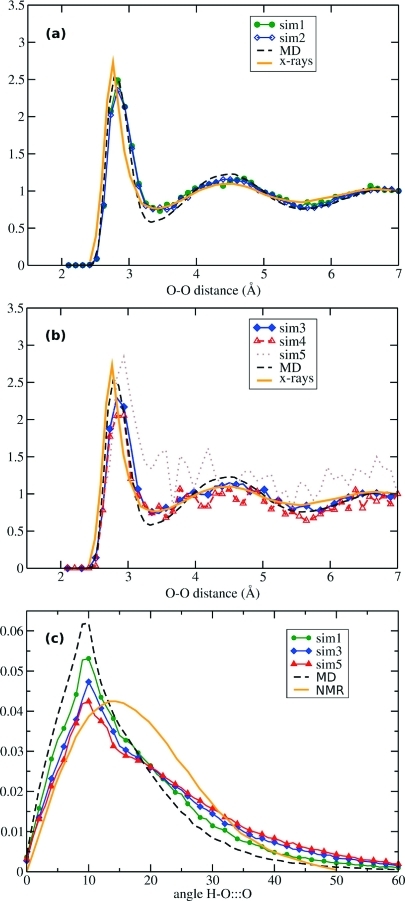
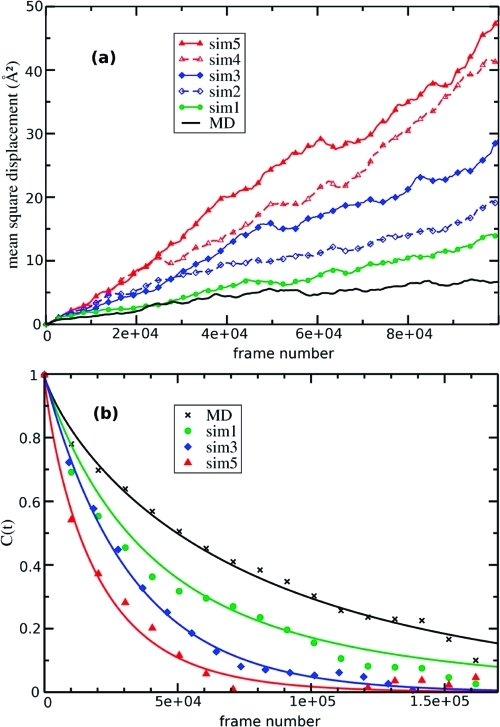
We have implemented the accelerated molecular dynamics approach (Hamelberg, D.; Mongan, J.; McCammon, J. A. J. Chem. Phys. 2004, 120 (24), 11919) in the framework of ab initio MD (AIMD). Using three simple examples, we demonstrate that accelerated AIMD (A-AIMD) can be used to accelerate solvent relaxation in AIMD simulations and facilitate the detection of reaction coordinates: (i) We show, for one cyclohexane molecule in the gas phase, that the method can be used to accelerate the rate of the chair-to-chair interconversion by a factor of ∼1 × 10(5), while allowing for the reconstruction of the correct canonical distribution of low-energy states; (ii) We then show, for a water box of 64 H(2)O molecules, that A-AIMD can also be used in the condensed phase to accelerate the sampling of water conformations, without affecting the structural properties of the solvent; and (iii) The method is then used to compute the potential of mean force (PMF) for the dissociation of Na-Cl in water, accelerating the convergence by a factor of ∼3-4 compared to conventional AIMD simulations.(2) These results suggest that A-AIMD is a useful addition to existing methods for enhanced conformational and phase-space sampling in solution. While the method does not make the use of collective variables superfluous, it also does not require the user to define a set of collective variables that can capture all the low-energy minima on the potential energy surface. This property may prove very useful when dealing with highly complex multidimensional systems that require a quantum mechanical treatment.
我们已在从头算分子动力学(AIMD)框架内实现了加速分子动力学方法(哈梅尔贝格,D.;蒙根,J.;麦卡蒙,J. A.《化学物理杂志》2004年,120(24),11919)。通过三个简单示例,我们证明加速AIMD(A - AIMD)可用于加速AIMD模拟中的溶剂弛豫并促进反应坐标的检测:(i)对于气相中的一个环己烷分子,我们表明该方法可用于将椅式到椅式的互变速率加速约1×10⁵倍,同时允许重建低能态的正确正则分布;(ii)然后,对于一个由64个H₂O分子组成的水盒,我们表明A - AIMD也可用于凝聚相以加速水构象的采样,而不影响溶剂的结构性质;(iii)该方法随后用于计算水中Na - Cl解离的平均力势(PMF),与传统AIMD模拟相比,收敛速度加快了约3 - 4倍。(2)这些结果表明,A - AIMD是现有用于增强溶液中构象和相空间采样方法的有益补充。虽然该方法并没有使集体变量变得多余,但它也不要求用户定义一组能够捕获势能面上所有低能极小值的集体变量。在处理需要量子力学处理的高度复杂多维系统时,这一特性可能会非常有用。