Center for Medical Statistics, Informatics and Intelligent Systems, Department for Biosimulation and Bioinformatics, Medical University of Vienna, Austria.
BMC Bioinformatics. 2011 Jun 17;12:241. doi: 10.1186/1471-2105-12-241.
The binding between the major histocompatibility complex and the presented peptide is an indispensable prerequisite for the adaptive immune response. There is a plethora of different in silico techniques for the prediction of the peptide binding affinity to major histocompatibility complexes. Most studies screen a set of peptides for promising candidates to predict possible T cell epitopes. In this study we ask the question vice versa: Which peptides do have highest binding affinities to a given major histocompatibility complex according to certain in silico scoring functions?
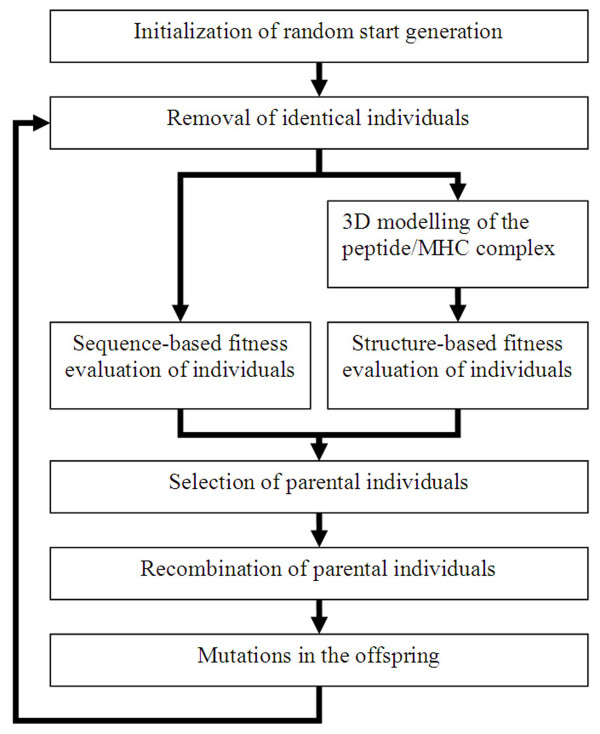
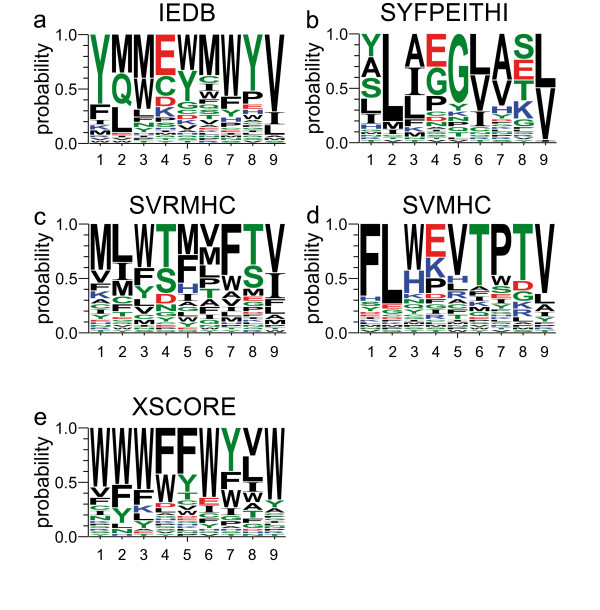
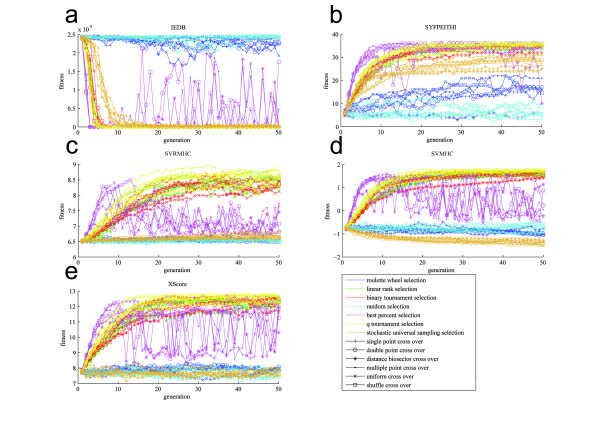
Since a full screening of all possible peptides is not feasible in reasonable runtime, we introduce a heuristic approach. We developed a framework for Genetic Algorithms to optimize peptides for the binding to major histocompatibility complexes. In an extensive benchmark we tested various operator combinations. We found that (1) selection operators have a strong influence on the convergence of the population while recombination operators have minor influence and (2) that five different binding prediction methods lead to five different sets of "optimal" peptides for the same major histocompatibility complex. The consensus peptides were experimentally verified as high affinity binders.
We provide a generalized framework to calculate sets of high affinity binders based on different previously published scoring functions in reasonable runtime. Furthermore we give insight into the different behaviours of operators and scoring functions of the Genetic Algorithm.
主要组织相容性复合体与呈递肽之间的结合是适应性免疫反应的不可或缺的前提条件。有大量不同的计算技术可用于预测肽与主要组织相容性复合体的结合亲和力。大多数研究筛选一组肽,以寻找有希望的候选肽来预测可能的 T 细胞表位。在这项研究中,我们反过来提出了一个问题:根据某些计算评分函数,哪些肽与给定的主要组织相容性复合体具有最高的结合亲和力?
由于在合理的时间内不可能对所有可能的肽进行全面筛选,因此我们引入了一种启发式方法。我们开发了一种用于遗传算法的框架,以优化与主要组织相容性复合体结合的肽。在广泛的基准测试中,我们测试了各种算子组合。我们发现:(1)选择算子对种群的收敛有很大的影响,而重组算子的影响较小;(2)五种不同的结合预测方法为同一主要组织相容性复合体产生了五种不同的“最佳”肽集。共识肽被实验证明为高亲和力结合物。
我们提供了一个通用的框架,可以在合理的时间内基于不同的先前发表的评分函数计算高亲和力结合物集。此外,我们深入了解了遗传算法算子和评分函数的不同行为。