Laboratory of Virology, Division of Infectious Diseases and Division of Laboratory Medicine, University of Geneva Hospitals, Geneva, Switzerland.
PLoS One. 2011;6(6):e21163. doi: 10.1371/journal.pone.0021163. Epub 2011 Jun 21.
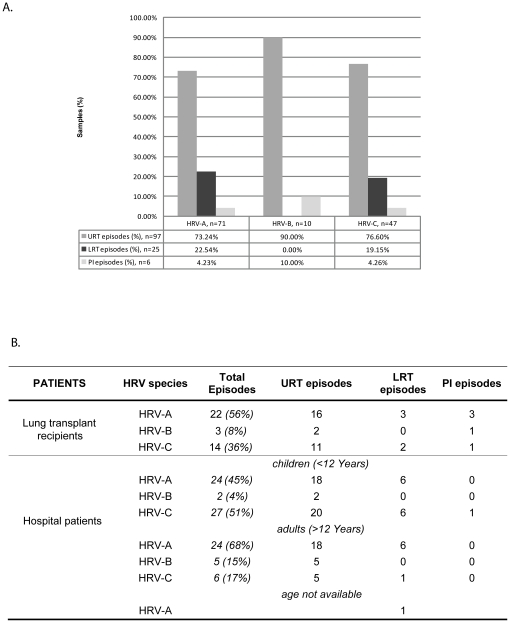
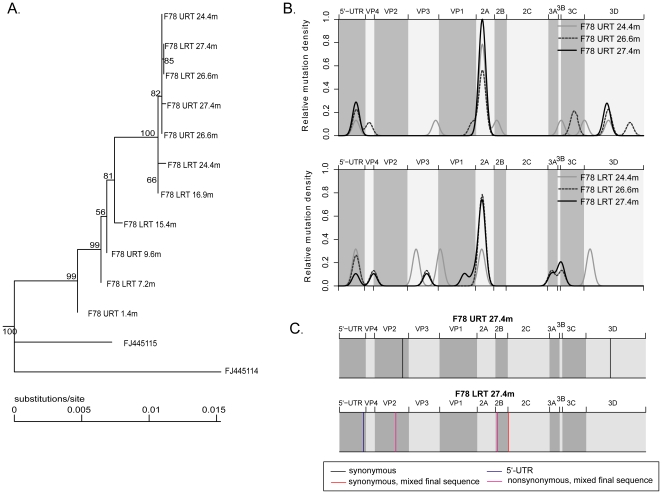
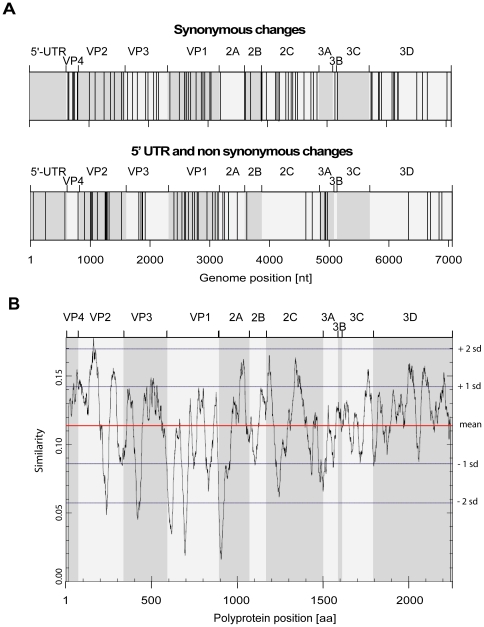
Routine screening of lung transplant recipients and hospital patients for respiratory virus infections allowed to identify human rhinovirus (HRV) in the upper and lower respiratory tracts, including immunocompromised hosts chronically infected with the same strain over weeks or months. Phylogenetic analysis of 144 HRV-positive samples showed no apparent correlation between a given viral genotype or species and their ability to invade the lower respiratory tract or lead to protracted infection. By contrast, protracted infections were found almost exclusively in immunocompromised patients, thus suggesting that host factors rather than the virus genotype modulate disease outcome, in particular the immune response. Complete genome sequencing of five chronic cases to study rhinovirus genome adaptation showed that the calculated mutation frequency was in the range observed during acute human infections. Analysis of mutation hot spot regions between specimens collected at different times or in different body sites revealed that non-synonymous changes were mostly concentrated in the viral capsid genes VP1, VP2 and VP3, independent of the HRV type. In an immunosuppressed lung transplant recipient infected with the same HRV strain for more than two years, both classical and ultra-deep sequencing of samples collected at different time points in the upper and lower respiratory tracts showed that these virus populations were phylogenetically indistinguishable over the course of infection, except for the last month. Specific signatures were found in the last two lower respiratory tract populations, including changes in the 5'UTR polypyrimidine tract and the VP2 immunogenic site 2. These results highlight for the first time the ability of a given rhinovirus to evolve in the course of a natural infection in immunocompromised patients and complement data obtained from previous experimental inoculation studies in immunocompetent volunteers.
对肺移植受者和住院患者进行呼吸道病毒感染常规筛查,可在上、下呼吸道中发现人类鼻病毒(HRV),包括慢性感染同一病毒株数周或数月的免疫功能低下宿主。对 144 份 HRV 阳性样本的系统进化分析表明,特定病毒基因型或种与其侵袭下呼吸道或导致持续感染的能力之间无明显相关性。相比之下,持续感染几乎仅见于免疫功能低下的患者,这表明宿主因素而非病毒基因型调节疾病结局,特别是免疫反应。对 5 例慢性感染病例进行完整基因组测序以研究鼻病毒基因组适应性的研究表明,计算出的突变频率处于急性人类感染期间观察到的范围内。对不同时间或不同身体部位采集的标本突变热点区域的分析表明,非同义变化主要集中在病毒衣壳基因 VP1、VP2 和 VP3 中,与 HRV 型无关。在一名感染同一 HRV 株超过两年的免疫抑制肺移植受者中,对上、下呼吸道不同时间点采集的样本进行经典测序和超深度测序显示,在感染过程中,除了最后一个月外,这些病毒群体在系统进化上无法区分。在下呼吸道的最后两个病毒群体中发现了特定的特征,包括 5'UTR 多嘧啶序列和 VP2 免疫原性位点 2 的变化。这些结果首次强调了特定鼻病毒在免疫功能低下患者自然感染过程中进化的能力,并补充了以前在免疫功能正常志愿者中进行的实验接种研究获得的数据。