Department of Plant Pathology, Kansas State University, 4024 Throckmorton Plant Sciences Center, Manhattan, KS 66506, USA.
BMC Genomics. 2011 Jul 7;12:352. doi: 10.1186/1471-2164-12-352.
Eight diverse sorghum (Sorghum bicolor L. Moench) accessions were subjected to short-read genome sequencing to characterize the distribution of single-nucleotide polymorphisms (SNPs). Two strategies were used for DNA library preparation. Missing SNP genotype data were imputed by local haplotype comparison. The effect of library type and genomic diversity on SNP discovery and imputation are evaluated.
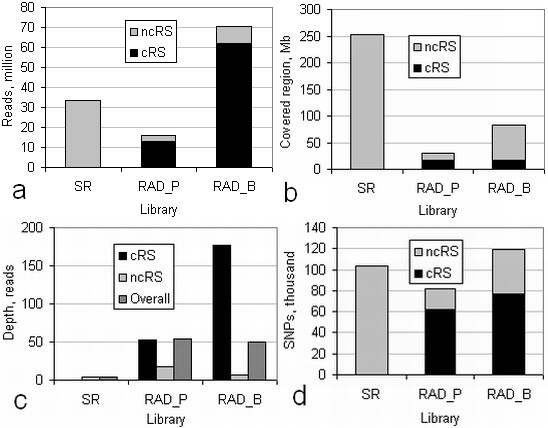
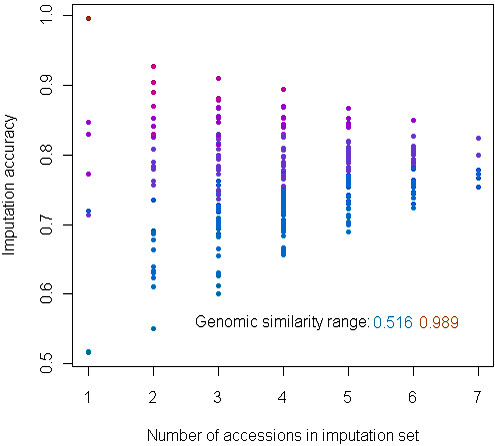
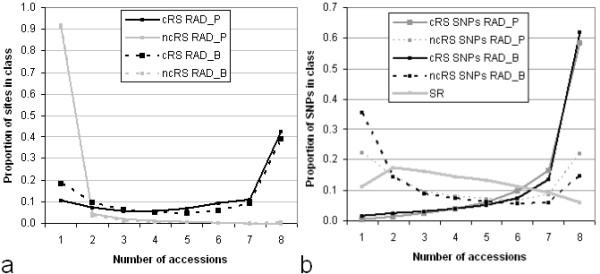
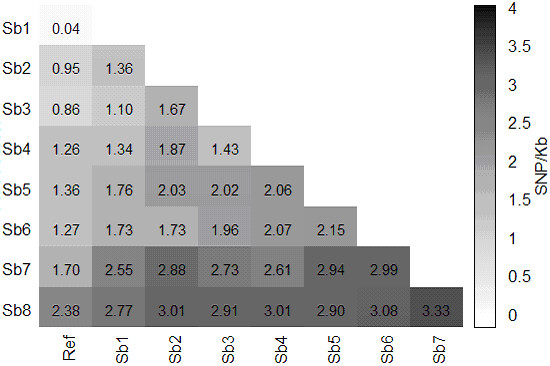
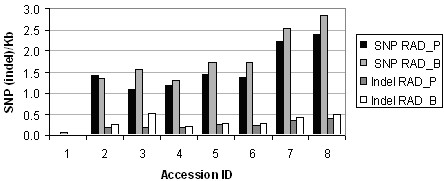
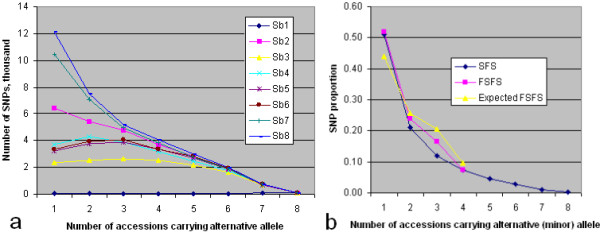
Alignment of eight genome equivalents (6 Gb) to the public reference genome revealed 283,000 SNPs at ≥82% confirmation probability. Sequencing from libraries constructed to limit sequencing to start at defined restriction sites led to genotyping 10-fold more SNPs in all 8 accessions, and correctly imputing 11% more missing data, than from semirandom libraries. The SNP yield advantage of the reduced-representation method was less than expected, since up to one fifth of reads started at noncanonical restriction sites and up to one third of restriction sites predicted in silico to yield unique alignments were not sampled at near-saturation. For imputation accuracy, the availability of a genomically similar accession in the germplasm panel was more important than panel size or sequencing coverage.
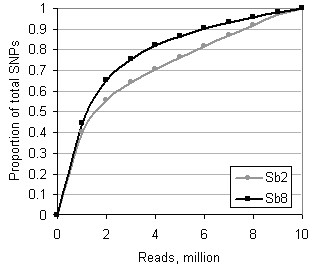
A sequence quantity of 3 million 50-base reads per accession using a BsrFI library would conservatively provide satisfactory genotyping of 96,000 sorghum SNPs. For most reliable SNP-genotype imputation in shallowly sequenced genomes, germplasm panels should consist of pairs or groups of genomically similar entries. These results may help in designing strategies for economical genotyping-by-sequencing of large numbers of plant accessions.
对 8 个不同的高粱(Sorghum bicolor L. Moench)品系进行短读长基因组测序,以研究单核苷酸多态性(SNP)的分布。采用了两种策略进行 DNA 文库制备。通过局部单倍型比较来推断缺失 SNP 基因型数据。评估了文库类型和基因组多样性对 SNP 发现和推断的影响。
将 8 个基因组当量(6 Gb)与公共参考基因组比对,在 ≥82%置信概率下发现了 283,000 个 SNP。从构建的文库中进行测序,限制测序从定义的限制位点开始,导致在所有 8 个品系中鉴定出的 SNP 数量增加了 10 倍,并且正确推断出的缺失数据增加了 11%,而从半随机文库中鉴定出的 SNP 数量则增加了 10 倍。减少代表性方法的 SNP 产量优势低于预期,因为多达五分之一的读取从非规范限制位点开始,多达三分之一的在硅中预测产生独特比对的限制位点在接近饱和的情况下没有被采样。对于推断准确性,种质资源库中是否存在基因组相似的品系比库大小或测序覆盖率更为重要。
每个品系使用 BsrFI 文库,每个品系使用 300 万个 50 碱基的读长,可保守地提供 96000 个高粱 SNP 的满意基因型鉴定。为了在浅测序基因组中实现最可靠的 SNP 基因型推断,种质资源库应包括基因组相似的品系对或群体。这些结果可能有助于设计经济高效的高通量测序策略,用于对大量植物品系进行基因分型。