Department of Computer Science, UC Davis, 1 Shields Ave., Davis, CA 95616, USA.
BMC Bioinformatics. 2011 Jul 14;12:287. doi: 10.1186/1471-2105-12-287.
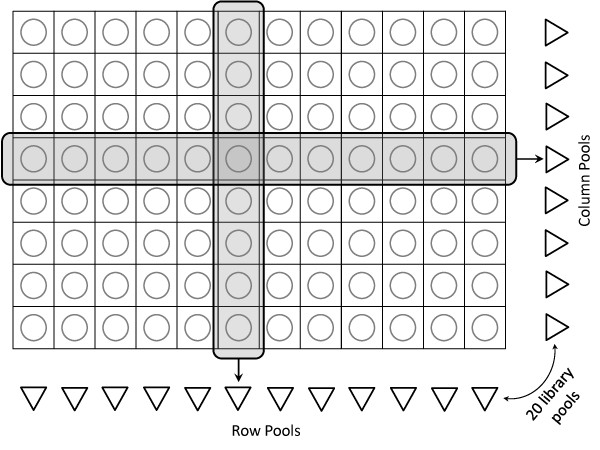
TILLING (Targeting induced local lesions IN genomes) is an efficient reverse genetics approach for detecting induced mutations in pools of individuals. Combined with the high-throughput of next-generation sequencing technologies, and the resolving power of overlapping pool design, TILLING provides an efficient and economical platform for functional genomics across thousands of organisms.
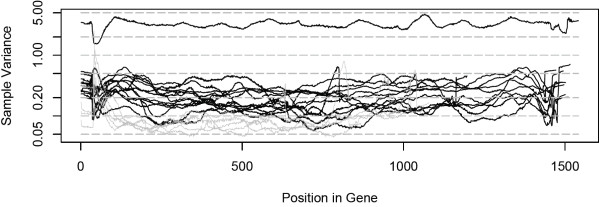
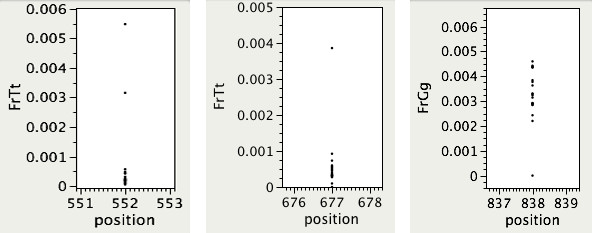
We propose a probabilistic method for calling TILLING-induced mutations, and their carriers, from high throughput sequencing data of overlapping population pools, where each individual occurs in two pools. We assign a probability score to each sequence position by applying Bayes' Theorem to a simplified binomial model of sequencing error and expected mutations, taking into account the coverage level. We test the performance of our method on variable quality, high-throughput sequences from wheat and rice mutagenized populations.
We show that our method effectively discovers mutations in large populations with sensitivity of 92.5% and specificity of 99.8%. It also outperforms existing SNP detection methods in detecting real mutations, especially at higher levels of coverage variability across sequenced pools, and in lower quality short reads sequence data. The implementation of our method is available from: http://www.cs.ucdavis.edu/filkov/CAMBa/.
TILLING(靶向诱导基因组局部突变)是一种有效的反向遗传学方法,用于检测个体群体中诱导的突变。结合高通量的下一代测序技术和重叠池设计的分辨率,TILLING 为数千种生物的功能基因组学提供了高效、经济的平台。
我们提出了一种从重叠群体池高通量测序数据中呼叫 TILLING 诱导突变及其携带者的概率方法,其中每个个体出现在两个池中。我们通过将贝叶斯定理应用于测序错误和预期突变的简化二项式模型,并考虑覆盖水平,为每个序列位置分配概率得分。我们在经过诱变的小麦和水稻群体的可变质量、高通量序列上测试了我们方法的性能。
我们表明,我们的方法在敏感性为 92.5%和特异性为 99.8%的情况下,有效地在大群体中发现突变。它还在检测真实突变方面优于现有的 SNP 检测方法,特别是在测序池之间的覆盖变异水平更高、低质量短读序列数据的情况下。我们的方法的实现可从以下网址获得:http://www.cs.ucdavis.edu/filkov/CAMBa/。