Department of Chemistry and Biochemistry , Ohio State University , Columbus , Ohio 43210 , United States.
Department of Chemistry/Biochemistry , Oberlin College , Oberlin , Ohio 44074 , United States.
J Phys Chem B. 2019 Aug 22;123(33):7103-7112. doi: 10.1021/acs.jpcb.9b04333. Epub 2019 Aug 14.
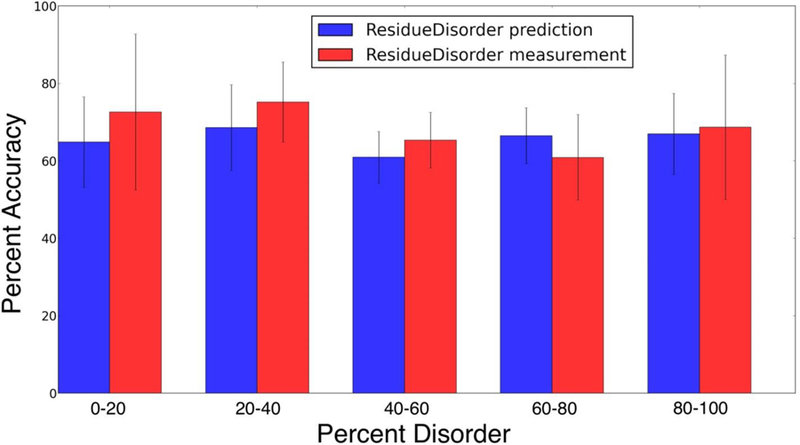
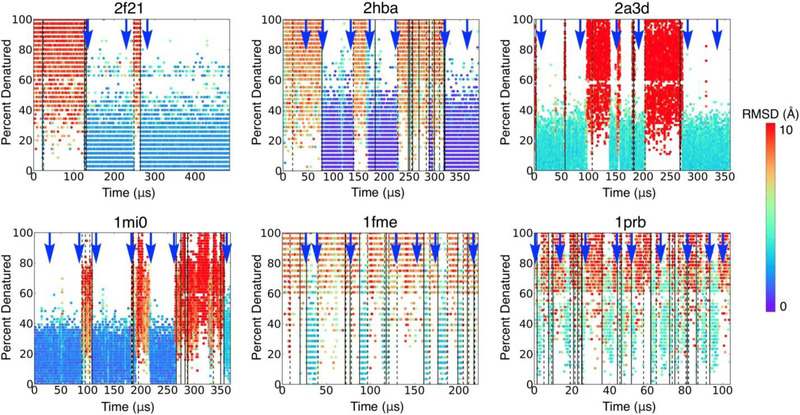
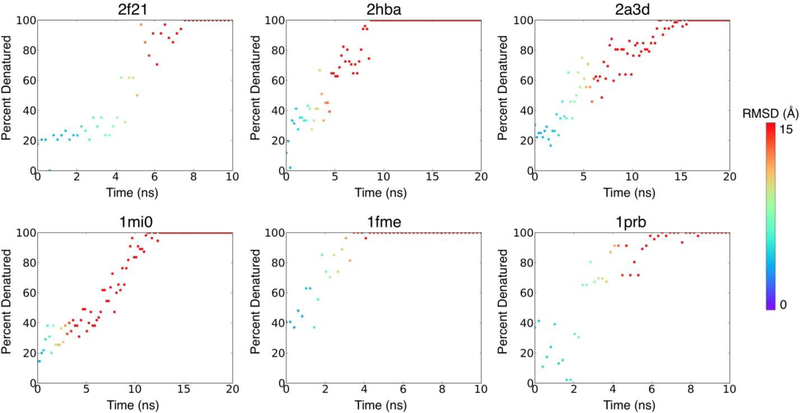
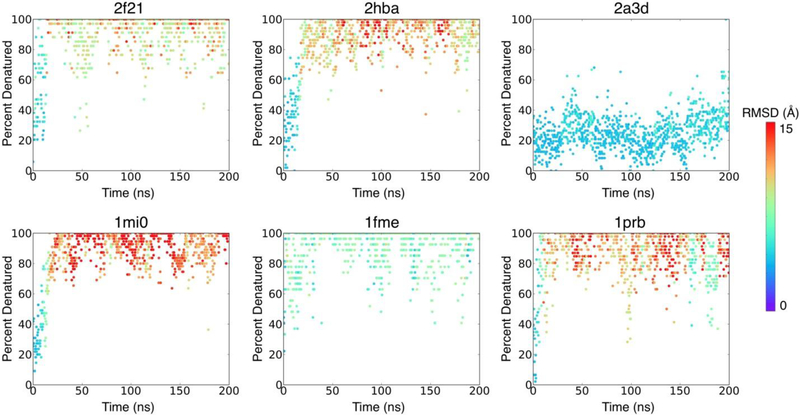
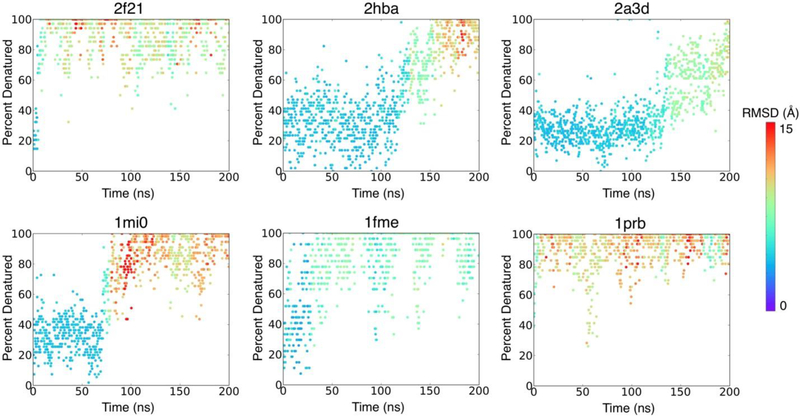
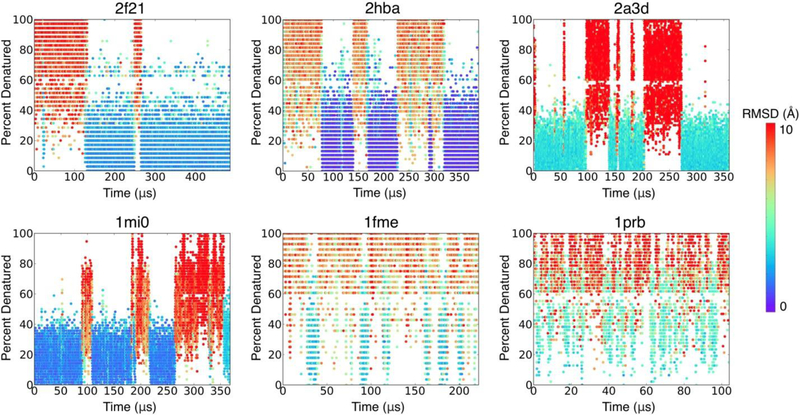
Many proteins contain regions of intrinsic disorder, not folding into unique, stable conformations. Numerous experimental methods have been developed to measure the disorder of all or select residues. In the absence of experimental data, computational methods are often utilized to identify these disordered regions and thus gain a better understanding of both structure and function. Many freely available computational methods have been developed to predict regions of intrinsic disorder from the primary sequence of a protein, including our recently developed Rosetta ResidueDisorder. While these methods are very useful, they are only designed to predict intrinsic disorder from the sequence. However, it would be useful to have a method that could also measure intrinsic disorder directly from structure. Such a method might also be used to identify changes in the structure of systems that can transition from folded to unfolded or vice versa, even systems that are not intrinsically disordered. Here we extended the capabilities of Rosetta ResidueDisorder to measure the intrinsic disorder from the coordinates of a single conformation of a protein. As a proof of principle, we show that ResidueDisorder can measure the intrinsic disorder from the coordinates with a higher accuracy (69.2%) than when predicted from sequence (65.4%) using a benchmark set of 229 proteins, showing that intrinsic disorder can be measured accurately from single structures over a large range of intrinsic disorder (0-100%). Additionally, we used ResidueDisorder to analyze unfolding trajectories of 12 fast-folding, nonintrinsically disordered proteins generated using molecular dynamics (MD), specifically steered MD (SMD), high-temperature MD, and accelerated MD (aMD) as well as long-time scale folding/unfolding trajectories. Using ResidueDisorder, a clear correlation between RMSD with respect to the native structure and measured fraction of denatured residues was observed. Finally, we introduced methods to predict folding/unfolding transitions as well as a native-like structure in the absence of a crystal structure from folding/unfolding MD trajectories. Rosetta ResidueDisorder is available as an application in the Rosetta software suite with the addition of new capabilities for directly identifying denatured regions and predicting events.
许多蛋白质包含内在无序区域,无法折叠成独特、稳定的构象。已经开发了许多实验方法来测量所有或选定残基的无序程度。在没有实验数据的情况下,通常利用计算方法来识别这些无序区域,从而更好地理解结构和功能。已经开发了许多免费的计算方法来从蛋白质的一级序列预测内在无序区域,包括我们最近开发的 Rosetta ResidueDisorder。虽然这些方法非常有用,但它们仅设计用于从序列预测内在无序。然而,拥有一种可以直接从结构测量内在无序的方法将是有用的。这种方法也可用于识别可以从折叠状态转变为展开状态或反之亦然的系统的结构变化,即使系统本身不是内在无序的。在这里,我们扩展了 Rosetta ResidueDisorder 的功能,以便从单个蛋白质构象的坐标测量内在无序。作为原理验证,我们表明 ResidueDisorder 可以比从序列预测(65.4%)更高的精度(69.2%)从坐标测量内在无序,使用 229 个蛋白质的基准集,这表明可以从大范围的内在无序(0-100%)的单个结构准确测量内在无序。此外,我们使用 ResidueDisorder 分析了使用分子动力学(MD)生成的 12 种快速折叠、非内在无序蛋白质的展开轨迹,具体为导向 MD(SMD)、高温 MD 和加速 MD(aMD)以及长时间尺度的折叠/展开轨迹。使用 ResidueDisorder,观察到与天然结构的均方根偏差(RMSD)和测量的变性残基分数之间存在明显的相关性。最后,我们引入了从折叠/展开 MD 轨迹预测折叠/展开转变以及无晶体结构的天然样结构的方法。Rosetta ResidueDisorder 可作为 Rosetta 软件套件中的应用程序使用,增加了直接识别变性区域和预测事件的新功能。