Bioinformatics Center, Institute for Chemical Research, Kyoto University, Gokasho, Uji, Japan.
PLoS One. 2011;6(7):e22281. doi: 10.1371/journal.pone.0022281. Epub 2011 Jul 29.
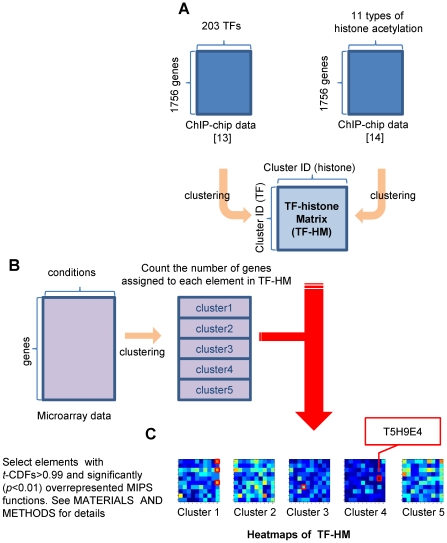
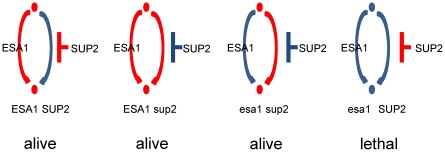
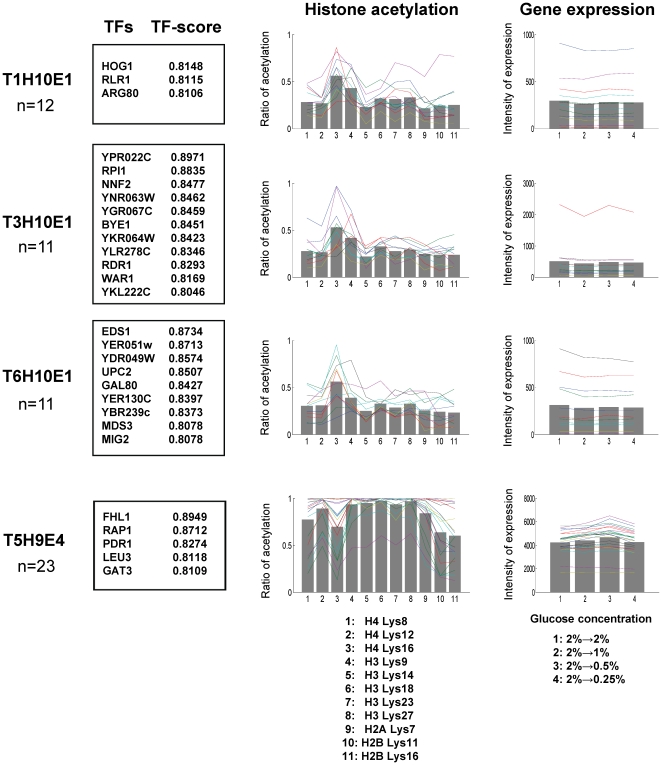
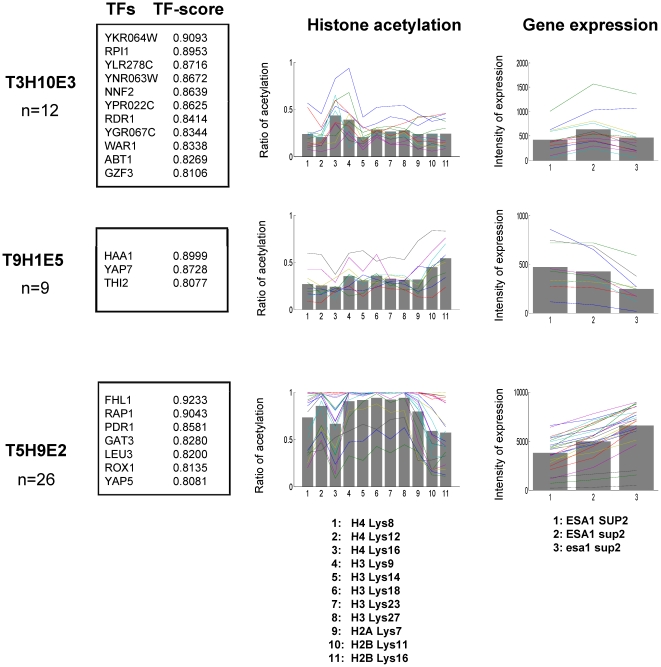
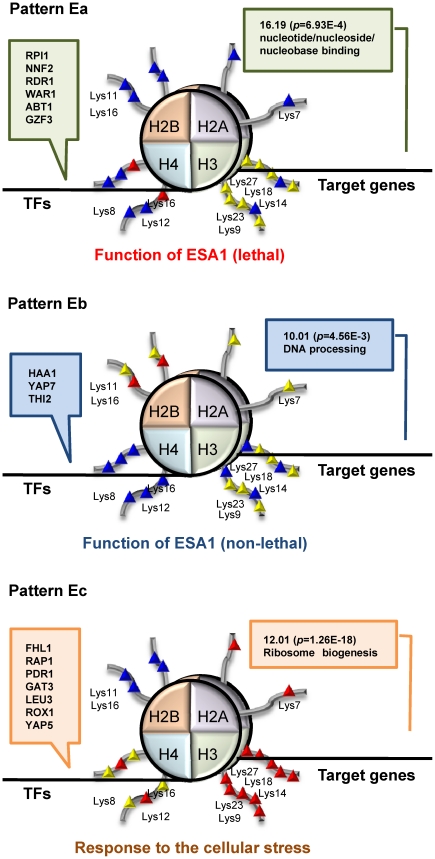
N-terminal tails of H2A, H2B, H3 and H4 histone families are subjected to posttranslational modifications that take part in transcriptional regulation mechanisms, such as transcription factor binding and gene expression. Regulation mechanisms under control of histone modification are important but remain largely unclear, despite of emerging datasets for comprehensive analysis of histone modification. In this paper, we focus on what we call genetic harmonious units (GHUs), which are co-occurring patterns among transcription factor binding, gene expression and histone modification. We present the first genome-wide approach that captures GHUs by combining ChIP-chip with microarray datasets from Saccharomyces cerevisiae. Our approach employs noise-robust soft clustering to select patterns which share the same preferences in transcription factor-binding, histone modification and gene expression, which are all currently implied to be closely correlated. The detected patterns are a well-studied acetylation of lysine 16 of H4 in glucose depletion as well as co-acetylation of five lysine residues of H3 with H4 Lys12 and H2A Lys7 responsible for ribosome biogenesis. Furthermore, our method further suggested the recognition of acetylated H4 Lys16 being crucial to histone acetyltransferase ESA1, whose essential role is still under controversy, from a microarray dataset on ESA1 and its bypass suppressor mutants. These results demonstrate that our approach allows us to provide clearer principles behind gene regulation mechanisms under histone modifications and detect GHUs further by applying to other microarray and ChIP-chip datasets. The source code of our method, which was implemented in MATLAB (http://www.mathworks.com/), is available from the supporting page for this paper: http://www.bic.kyoto-u.ac.jp/pathway/natsume/hm_detector.htm.
组蛋白 H2A、H2B、H3 和 H4 家族的 N-末端尾部会发生翻译后修饰,这些修饰参与转录调控机制,如转录因子结合和基因表达。尽管有综合分析组蛋白修饰的新兴数据集,但受组蛋白修饰控制的调控机制仍然非常重要,但在很大程度上仍不清楚。在本文中,我们专注于我们所谓的遗传协调单元 (GHU),这是转录因子结合、基因表达和组蛋白修饰之间共同出现的模式。我们提出了一种全基因组方法,通过将 ChIP-chip 与来自酿酒酵母的微阵列数据集相结合,来捕获 GHU。我们的方法采用抗噪软聚类来选择具有相同转录因子结合、组蛋白修饰和基因表达偏好的模式,这些模式目前都被认为是密切相关的。检测到的模式是葡萄糖耗竭时组蛋白 H4 赖氨酸 16 的乙酰化以及与组蛋白 H3 的赖氨酸 5 的共乙酰化,这与核糖体生物发生有关,而组蛋白 H4 赖氨酸 12 和 H2A 赖氨酸 7 负责核糖体生物发生。此外,我们的方法还进一步表明,从 ESA1 及其旁路抑制突变体的微阵列数据集中,识别乙酰化的 H4 赖氨酸 16 对组蛋白乙酰转移酶 ESA1 至关重要,而 ESA1 的重要作用仍存在争议。这些结果表明,我们的方法使我们能够提供更清晰的组蛋白修饰下基因调控机制的原理,并通过应用于其他微阵列和 ChIP-chip 数据集来检测 GHU。我们的方法的源代码是用 MATLAB 编写的 (http://www.mathworks.com/),可从本文的支持页面获得:http://www.bic.kyoto-u.ac.jp/pathway/natsume/hm_detector.htm。