Department of Ecology and Evolutionary Biology, Brown University, Providence, Rhode Island, United States of America.
PLoS One. 2011;6(7):e22953. doi: 10.1371/journal.pone.0022953. Epub 2011 Jul 29.
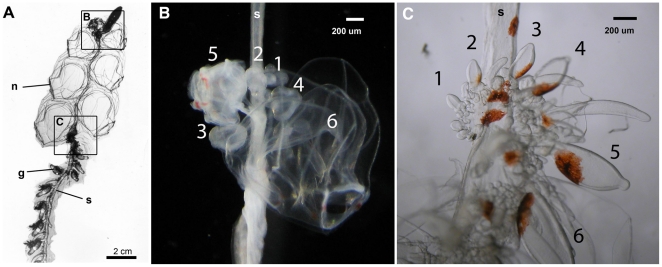
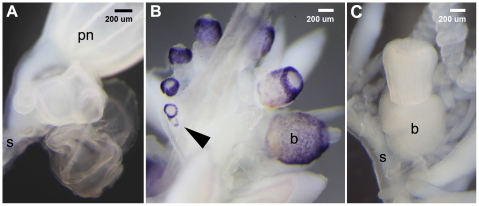
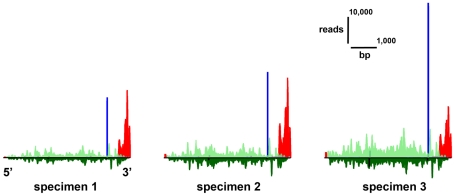
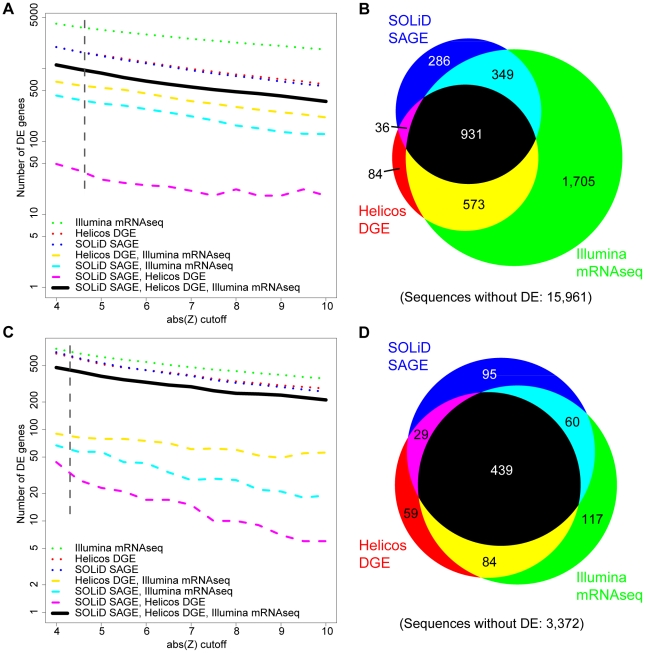
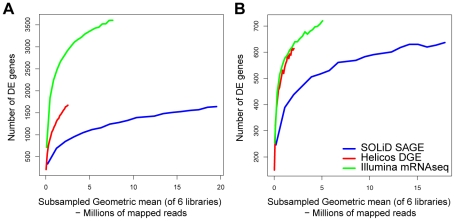
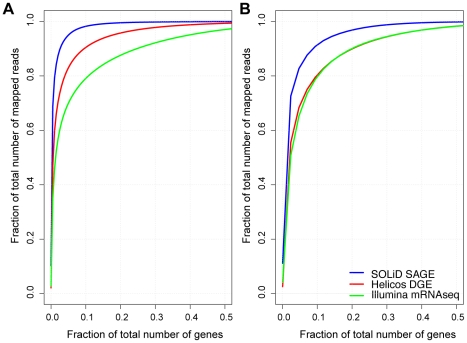
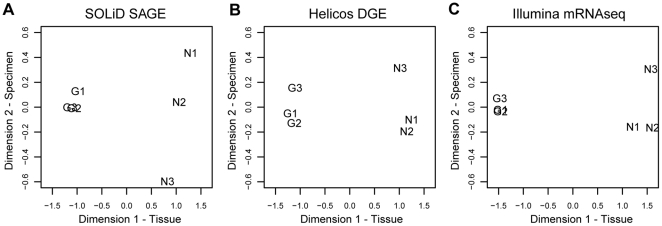
We investigated differential gene expression between functionally specialized feeding polyps and swimming medusae in the siphonophore Nanomia bijuga (Cnidaria) with a hybrid long-read/short-read sequencing strategy. We assembled a set of partial gene reference sequences from long-read data (Roche 454), and generated short-read sequences from replicated tissue samples that were mapped to the references to quantify expression. We collected and compared expression data with three short-read expression workflows that differ in sample preparation, sequencing technology, and mapping tools. These workflows were Illumina mRNA-Seq, which generates sequence reads from random locations along each transcript, and two tag-based approaches, SOLiD SAGE and Helicos DGE, which generate reads from particular tag sites. Differences in expression results across workflows were mostly due to the differential impact of missing data in the partial reference sequences. When all 454-derived gene reference sequences were considered, Illumina mRNA-Seq detected more than twice as many differentially expressed (DE) reference sequences as the tag-based workflows. This discrepancy was largely due to missing tag sites in the partial reference that led to false negatives in the tag-based workflows. When only the subset of reference sequences that unambiguously have tag sites was considered, we found broad congruence across workflows, and they all identified a similar set of DE sequences. Our results are promising in several regards for gene expression studies in non-model organisms. First, we demonstrate that a hybrid long-read/short-read sequencing strategy is an effective way to collect gene expression data when an annotated genome sequence is not available. Second, our replicated sampling indicates that expression profiles are highly consistent across field-collected animals in this case. Third, the impacts of partial reference sequences on the ability to detect DE can be mitigated through workflow choice and deeper reference sequencing.
我们采用混合长读/短读测序策略,研究了管水母目(刺胞动物门)Nanomia bijuga 中功能特化的摄食水螅体和游动水母体之间的差异基因表达。我们从长读数据(罗氏 454)组装了一组部分基因参考序列,并从重复的组织样本生成短读序列,将其映射到参考序列以定量表达。我们收集并比较了三个短读表达工作流程的表达数据,这些工作流程在样本制备、测序技术和映射工具方面有所不同。这些工作流程包括从每个转录本的随机位置生成序列读段的 Illumina mRNA-Seq,以及两种基于标签的方法 SOLiD SAGE 和 Helicos DGE,它们从特定的标签位点生成读段。工作流程之间的表达结果差异主要归因于部分参考序列中缺失数据的不同影响。当考虑所有 454 衍生的基因参考序列时,Illumina mRNA-Seq 检测到的差异表达(DE)参考序列数量是基于标签的工作流程的两倍多。这种差异主要是由于部分参考中缺失的标签位点导致基于标签的工作流程中出现假阴性。当只考虑具有明确标签位点的参考序列子集时,我们发现各工作流程之间存在广泛的一致性,它们都鉴定出了一组相似的 DE 序列。我们的研究结果在多个方面为非模式生物的基因表达研究提供了有希望的结果。首先,我们证明了在没有注释基因组序列的情况下,混合长读/短读测序策略是收集基因表达数据的有效方法。其次,我们的重复采样表明,在这种情况下,来自野外采集的动物的表达谱高度一致。第三,通过工作流程选择和更深的参考测序,可以减轻部分参考序列对检测 DE 的能力的影响。