Department of Physics, University of Alabama, Birmingham, AL 35294, USA.
BMC Bioinformatics. 2011 Aug 31;12:359. doi: 10.1186/1471-2105-12-359.
Bayesian Network (BN) is a powerful approach to reconstructing genetic regulatory networks from gene expression data. However, expression data by itself suffers from high noise and lack of power. Incorporating prior biological knowledge can improve the performance. As each type of prior knowledge on its own may be incomplete or limited by quality issues, integrating multiple sources of prior knowledge to utilize their consensus is desirable.
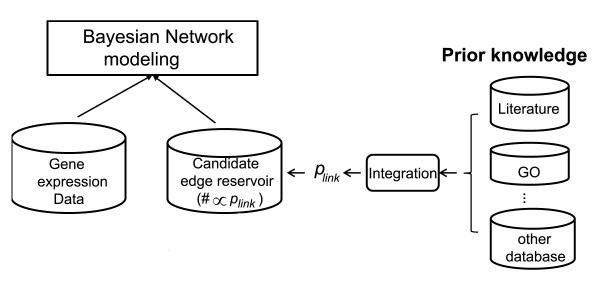
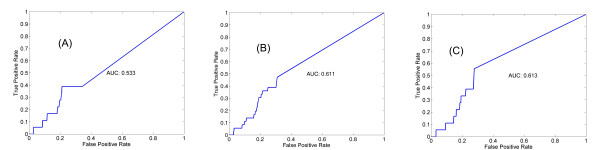
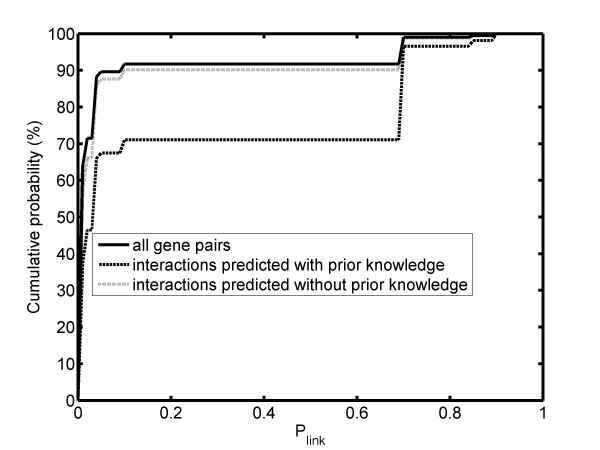
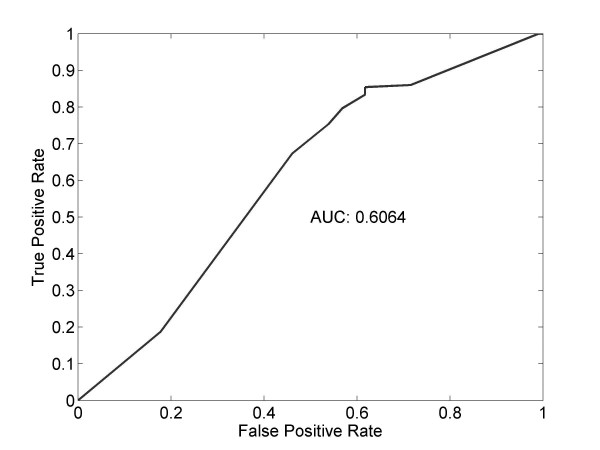
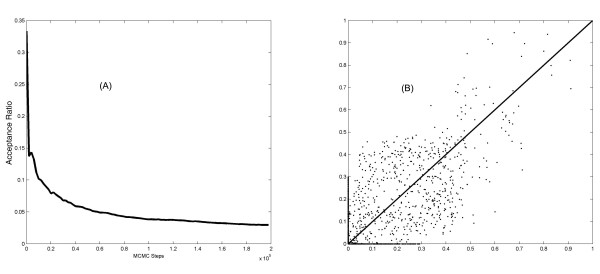
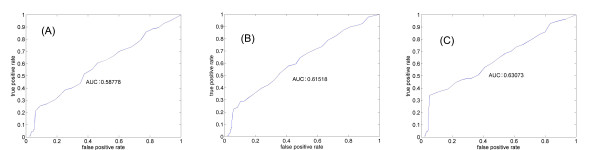
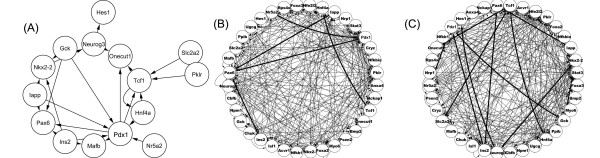
We introduce a new method to incorporate the quantitative information from multiple sources of prior knowledge. It first uses the Naïve Bayesian classifier to assess the likelihood of functional linkage between gene pairs based on prior knowledge. In this study we included cocitation in PubMed and schematic similarity in Gene Ontology annotation. A candidate network edge reservoir is then created in which the copy number of each edge is proportional to the estimated likelihood of linkage between the two corresponding genes. In network simulation the Markov Chain Monte Carlo sampling algorithm is adopted, and samples from this reservoir at each iteration to generate new candidate networks. We evaluated the new algorithm using both simulated and real gene expression data including that from a yeast cell cycle and a mouse pancreas development/growth study. Incorporating prior knowledge led to a ~2 fold increase in the number of known transcription regulations recovered, without significant change in false positive rate. In contrast, without the prior knowledge BN modeling is not always better than a random selection, demonstrating the necessity in network modeling to supplement the gene expression data with additional information.
our new development provides a statistical means to utilize the quantitative information in prior biological knowledge in the BN modeling of gene expression data, which significantly improves the performance.
贝叶斯网络(BN)是一种从基因表达数据中重建遗传调控网络的强大方法。然而,表达数据本身存在高噪声和缺乏信息的问题。结合先验生物学知识可以提高性能。由于每种先验知识本身可能不完整或受到质量问题的限制,因此整合多种来源的先验知识以利用它们的共识是可取的。
我们介绍了一种新的方法来整合来自多种来源的先验知识的定量信息。它首先使用朴素贝叶斯分类器根据先验知识评估基因对之间功能关联的可能性。在本研究中,我们包括 PubMed 中的共引和 Gene Ontology 注释中的示意图相似性。然后创建一个候选网络边缘库,其中每个边缘的副本数与两个对应基因之间链接的估计可能性成正比。在网络模拟中,采用马尔可夫链蒙特卡罗抽样算法,从该库中在每次迭代时采样以生成新的候选网络。我们使用模拟和真实基因表达数据(包括酵母细胞周期和小鼠胰腺发育/生长研究的数据)评估了新算法。结合先验知识可将已知转录调控的数量增加约 2 倍,而假阳性率没有显著变化。相比之下,如果没有先验知识,BN 建模并不总是优于随机选择,这表明在网络建模中需要用额外的信息来补充基因表达数据。
我们的新方法为利用 BN 对基因表达数据进行建模时的先验生物学知识中的定量信息提供了一种统计手段,显著提高了性能。