Waisman Center, University of Wisconsin-Madison, Madison, Wisconsin, United States of America.
PLoS One. 2011;6(9):e25255. doi: 10.1371/journal.pone.0025255. Epub 2011 Sep 26.
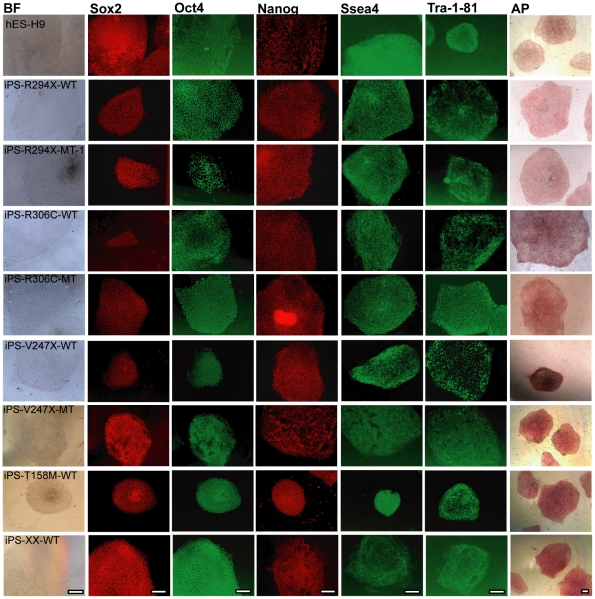
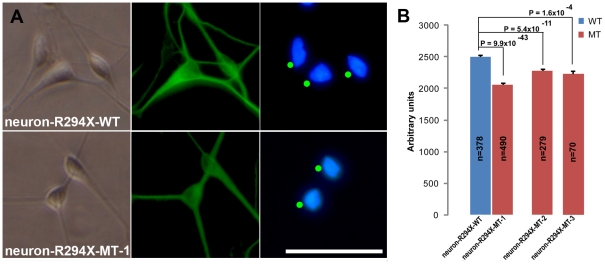
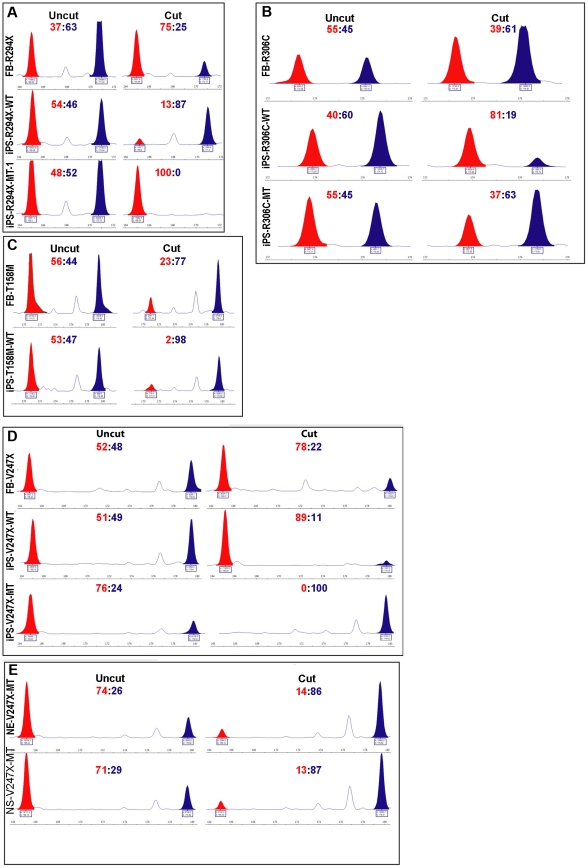
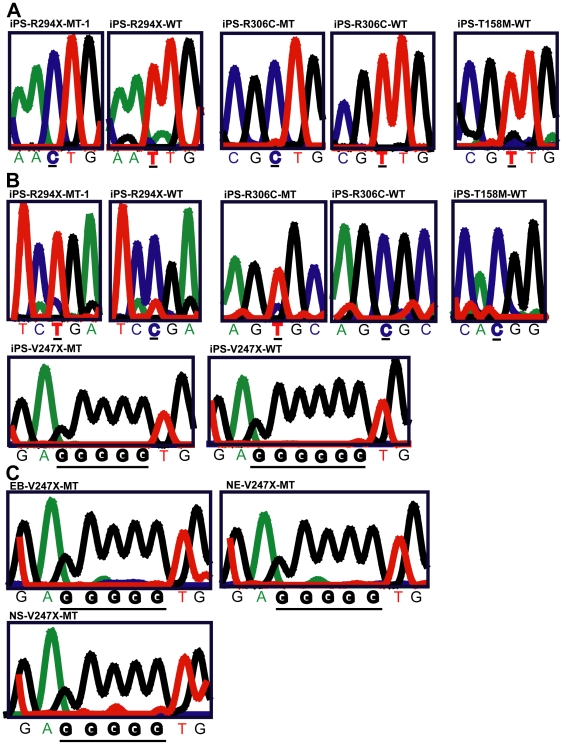
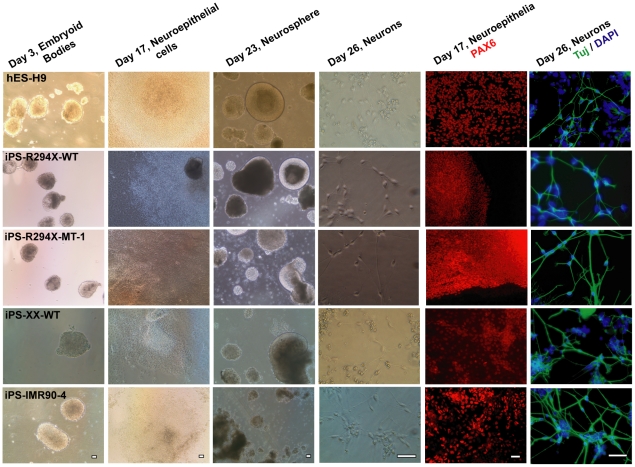
Rett syndrome (RTT) is an autism spectrum developmental disorder caused by mutations in the X-linked methyl-CpG binding protein 2 (MECP2) gene. Excellent RTT mouse models have been created to study the disease mechanisms, leading to many important findings with potential therapeutic implications. These include the identification of many MeCP2 target genes, better understanding of the neurobiological consequences of the loss- or mis-function of MeCP2, and drug testing in RTT mice and clinical trials in human RTT patients. However, because of potential differences in the underlying biology between humans and common research animals, there is a need to establish cell culture-based human models for studying disease mechanisms to validate and expand the knowledge acquired in animal models. Taking advantage of the nonrandom pattern of X chromosome inactivation in female induced pluripotent stem cells (iPSC), we have generated isogenic pairs of wild type and mutant iPSC lines from several female RTT patients with common and rare RTT mutations. R294X (arginine 294 to stop codon) is a common mutation carried by 5-6% of RTT patients. iPSCs carrying the R294X mutation has not been studied. We differentiated three R294X iPSC lines and their isogenic wild type control iPSC into neurons with high efficiency and consistency, and observed characteristic RTT pathology in R294X neurons. These isogenic iPSC lines provide unique resources to the RTT research community for studying disease pathology, screening for novel drugs, and testing toxicology.
雷特综合征(RTT)是一种由 X 连锁甲基化CpG 结合蛋白 2(MECP2)基因突变引起的自闭症谱系发育障碍。已经创建了出色的 RTT 小鼠模型来研究疾病机制,这导致了许多具有潜在治疗意义的重要发现。这些发现包括鉴定出许多 MeCP2 靶基因、更好地理解 MeCP2 缺失或功能异常的神经生物学后果,以及在 RTT 小鼠中进行药物测试和在人类 RTT 患者中进行临床试验。然而,由于人类和常见研究动物之间潜在的生物学差异,因此需要建立基于细胞培养的人类模型来研究疾病机制,以验证和扩展在动物模型中获得的知识。我们利用女性诱导多能干细胞(iPSC)中 X 染色体失活的非随机模式,从几个患有常见和罕见 RTT 突变的女性 RTT 患者中生成了野生型和突变型 iPSC 系的同基因对。R294X(精氨酸 294 到终止密码子)是 5-6%的 RTT 患者携带的常见突变。携带 R294X 突变的 iPSC 尚未进行研究。我们将三个 R294X iPSC 系及其同基因野生型对照 iPSC 高效且一致地分化为神经元,并在 R294X 神经元中观察到典型的 RTT 病理学。这些同基因 iPSC 系为 RTT 研究社区提供了独特的资源,可用于研究疾病病理学、筛选新药物和测试毒理学。