Baumler David J, Peplinski Roman G, Reed Jennifer L, Glasner Jeremy D, Perna Nicole T
Genome Center of Wisconsin, University of Wisconsin-Madison, Madison, Wisconsin, USA.
BMC Syst Biol. 2011 Nov 1;5:182. doi: 10.1186/1752-0509-5-182.
Despite the availability of numerous complete genome sequences from E. coli strains, published genome-scale metabolic models exist only for two commensal E. coli strains. These models have proven useful for many applications, such as engineering strains for desired product formation, and we sought to explore how constructing and evaluating additional metabolic models for E. coli strains could enhance these efforts.
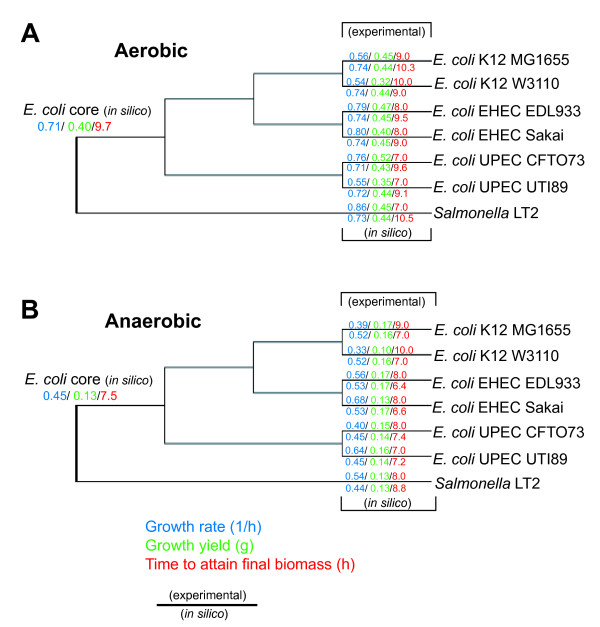
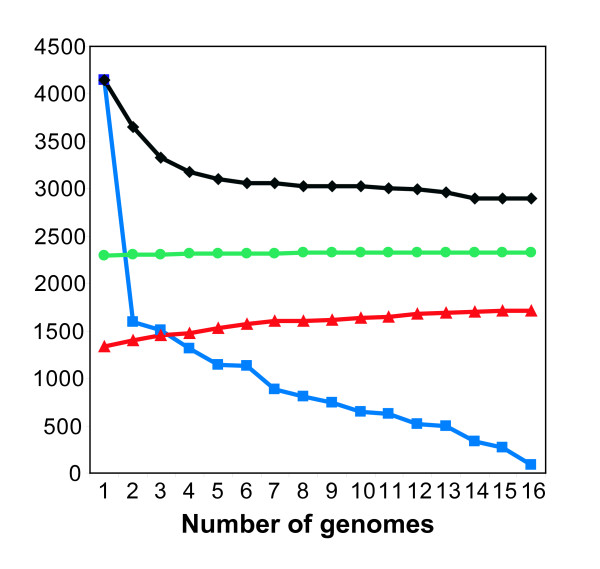
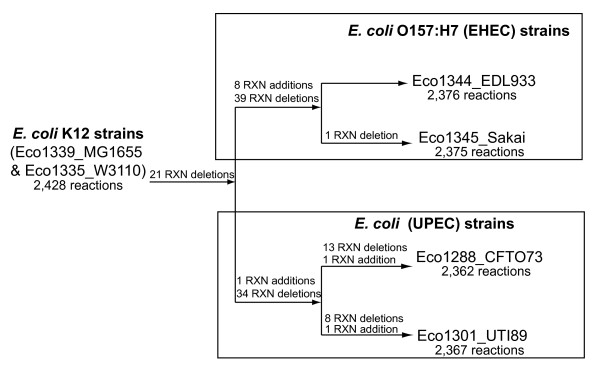
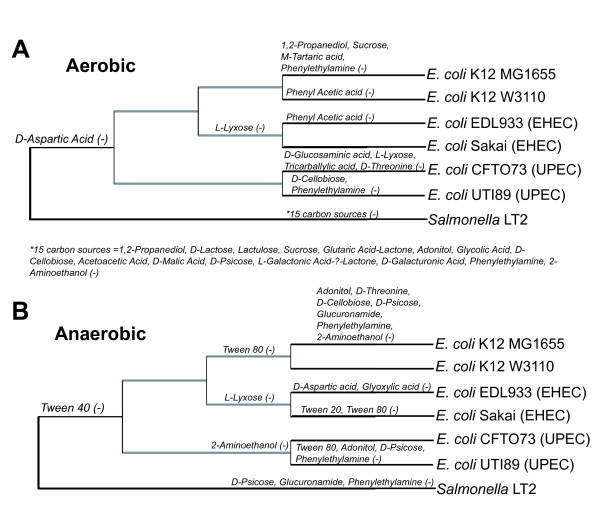
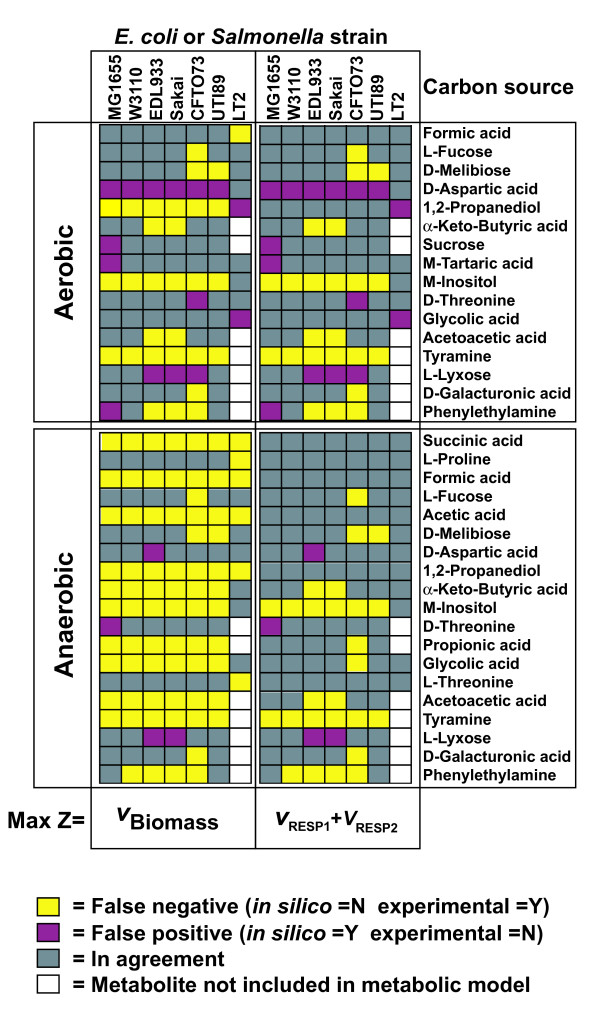
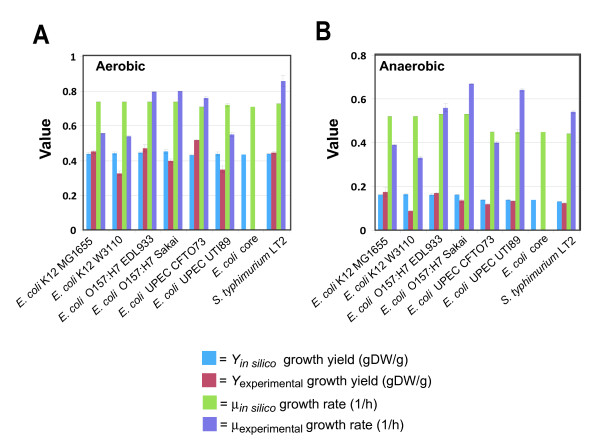
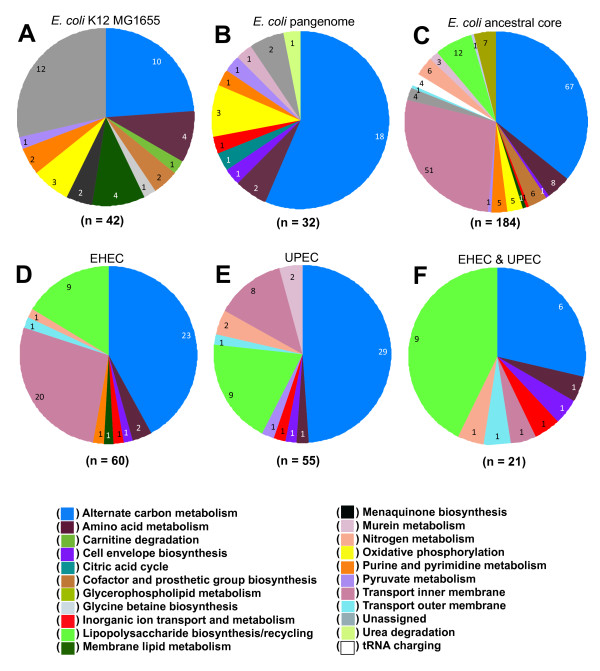
We used the genomic information from 16 E. coli strains to generate an E. coli pangenome metabolic network by evaluating their collective 76,990 ORFs. Each of these ORFs was assigned to one of 17,647 ortholog groups including ORFs associated with reactions in the most recent metabolic model for E. coli K-12. For orthologous groups that contain an ORF already represented in the MG1655 model, the gene to protein to reaction associations represented in this model could then be easily propagated to other E. coli strain models. All remaining orthologous groups were evaluated to see if new metabolic reactions could be added to generate a pangenome-scale metabolic model (iEco1712_pan). The pangenome model included reactions from a metabolic model update for E. coli K-12 MG1655 (iEco1339_MG1655) and enabled development of five additional strain-specific genome-scale metabolic models. These additional models include a second K-12 strain (iEco1335_W3110) and four pathogenic strains (two enterohemorrhagic E. coli O157:H7 and two uropathogens). When compared to the E. coli K-12 models, the metabolic models for the enterohemorrhagic (iEco1344_EDL933 and iEco1345_Sakai) and uropathogenic strains (iEco1288_CFT073 and iEco1301_UTI89) contained numerous lineage-specific gene and reaction differences. All six E. coli models were evaluated by comparing model predictions to carbon source utilization measurements under aerobic and anaerobic conditions, and to batch growth profiles in minimal media with 0.2% (w/v) glucose. An ancestral genome-scale metabolic model based on conserved ortholog groups in all 16 E. coli genomes was also constructed, reflecting the conserved ancestral core of E. coli metabolism (iEco1053_core). Comparative analysis of all six strain-specific E. coli models revealed that some of the pathogenic E. coli strains possess reactions in their metabolic networks enabling higher biomass yields on glucose. Finally the lineage-specific metabolic traits were compared to the ancestral core model predictions to derive new insight into the evolution of metabolism within this species.
Our findings demonstrate that a pangenome-scale metabolic model can be used to rapidly construct additional E. coli strain-specific models, and that quantitative models of different strains of E. coli can accurately predict strain-specific phenotypes. Such pangenome and strain-specific models can be further used to engineer metabolic phenotypes of interest, such as designing new industrial E. coli strains.
尽管已有众多大肠杆菌菌株的完整基因组序列,但已发表的全基因组规模代谢模型仅针对两种共生大肠杆菌菌株。这些模型已被证明在许多应用中有用,例如改造菌株以形成所需产物,我们试图探索构建和评估更多大肠杆菌菌株的代谢模型如何能加强这些工作。
我们利用16种大肠杆菌菌株的基因组信息,通过评估它们总共76,990个开放阅读框(ORF),生成了一个大肠杆菌泛基因组代谢网络。这些ORF中的每一个都被分配到17,647个直系同源组中的一个,包括与大肠杆菌K-12最新代谢模型中的反应相关的ORF。对于那些在MG1655模型中已有代表的ORF所在的直系同源组,该模型中基因到蛋白质再到反应的关联随后可以很容易地传播到其他大肠杆菌菌株模型中。对所有其余的直系同源组进行评估,以确定是否可以添加新的代谢反应来生成一个泛基因组规模的代谢模型(iEco1712_pan)。该泛基因组模型包括来自大肠杆菌K-12 MG1655代谢模型更新版(iEco1339_MG1655)的反应,并使得能够开发另外五个菌株特异性的全基因组规模代谢模型。这些额外的模型包括第二个K-12菌株(iEco1335_W3110)和四个致病菌株(两个肠出血性大肠杆菌O157:H7和两个尿路致病菌)。与大肠杆菌K-12模型相比,肠出血性(iEco1344_EDL933和iEco1345_Sakai)和尿路致病菌株(iEco1288_CFT073和iEco1301_UTI89)的代谢模型包含许多谱系特异性的基因和反应差异。通过将模型预测与需氧和厌氧条件下的碳源利用测量结果以及在含有0.2%(w/v)葡萄糖的基本培养基中的分批生长曲线进行比较,对所有六个大肠杆菌模型进行了评估。还构建了一个基于所有16个大肠杆菌基因组中保守直系同源组的祖先全基因组规模代谢模型,反映了大肠杆菌代谢的保守祖先核心(iEco1053_core)。对所有六个菌株特异性大肠杆菌模型的比较分析表明,一些致病性大肠杆菌菌株在其代谢网络中具有能够在葡萄糖上产生更高生物量产量的反应。最后,将谱系特异性代谢特征与祖先核心模型预测进行比较,以获得对该物种内代谢进化的新见解。
我们的研究结果表明,泛基因组规模的代谢模型可用于快速构建更多大肠杆菌菌株特异性模型,并且不同大肠杆菌菌株的定量模型可以准确预测菌株特异性表型。这种泛基因组和菌株特异性模型可进一步用于改造感兴趣的代谢表型,例如设计新的工业大肠杆菌菌株。